REVIEW ARTICLE
Wnt/β-Catenin Signalling during Liver Metabolism, Chronic Liver Disease and Hepatocarcinogenesis
Gayatri D. Shirolkar1*, Sara Pasic1*, Jully Gogoi-Tiwari1, Manoj K. Bhat2, John K. Olynyk3,4, Arun Dharmarajan1, Janina E. E. Tirnitz-Parker1,5
1School of Pharmacy and Biomedical Sciences and Curtin Health Innovation Research Institute, Curtin University, Bentley, WA, Australia; 2National Centre for Cell Science, Savitribai Phule Pune University Campus, Ganeshkhind, Pune, India; 3Fremantle and Fiona Stanley Hospitals, Perth, WA, Australia; 4School of Medical and Health Sciences, Edith Cowan University, Joondalup, WA, Australia; 5School of Medicine and Pharmacology, University of Western Australia, Fremantle, WA, Australia
*. These authors contributed equally to this manuscript.
Abstract
Chronic liver diseases (CLDs) are increasing in prevalence and their end-stage complications, namely, cirrhosis, liver failure and hepatocellular carcinoma represent major global challenges. The most common initiators of progressive CLD are viral hepatitis and long-term alcohol abuse as well as steatosis and steatohepatitis. Irrespective of the underlying aetiology, a common feature of CLD is the formation of hepatic ductular reactions, involving the proliferation of liver progenitor cells (LPCs) and their signalling to fibrosis-driving hepatic stellate cells. The Wnt/β-catenin pathway has been found to regulate development, stemness and differentiation, and alterations in its activity have been associated with tumour development. Recent data highlight the role of Wnt/β-catenin signalling in hepatic metabolism, steatosis and cancer, and suggest targeting of this pathway as a promising molecular strategy to potentially inhibit CLD progression and hepatocarcinogenesis.
Keywords: chronic liver disease; hepatocellular carcinoma; liver progenitor cells; metabolic syndrome; Wnt/β-catenin signalling;
Received: 26 January 2018;
Accepted: 20 February 2018;
Published: 15 March 2018
Author for correspondence: Janina E. E. Tirnitz-Parker, School of Pharmacy and Biomedical Sciences and Curtin Health Innovation Research Institute, Curtin University, Kent Street, Bentley, 6102, WA, Australia. Email:
N.Tirnitz-Parker@curtin.edu.auHow to cite: Shirolkar GD et al. Wnt/β-catenin signalling during liver metabolism, chronic liver disease and hepatocarcinogenesis. J Ren Hepat Disord. 2018;2(1):1–9.
Doi:
http://dx.doi.org/10.15586/jrenhep.2018.29Copyright: Shirolkar GD et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0
Introduction
Chronic liver disease (CLD) has become one of the most common causes of death globally with an estimated 1.03 million deaths per year, as reported in 2017. Excessive alcohol consumption, viral hepatitis and hepatic steatosis are the most prevalent risk factors for the initiation and progression of CLD (1). A UK report stated that standardised CLD mortality rates have increased by 400% since 1970, reflecting its growing burden and major challenge for global health (2). End-stage complications of CLD include cirrhosis, liver failure and malignancies, with hepatocellular carcinoma constituting 85–90% of all liver cancers (3). Current therapy options for hepatocellular carcinoma include surgical resection, radiofrequency ablation, transarterial chemoembolisation and orthotopic liver transplantation. The multikinase inhibitors sorafenib and regorafenib are the only systemic treatments with proven survival benefits and they prolong the life expectancy of patients by 2 to 3 months (4). Immune-based approaches, including targeting of the immune checkpoint inhibitors programmed cell death (PD-1), programmed cell death ligand 1 (PD-L1) or cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), represent novel, promising therapeutic strategies to prevent or treat hepatocellular carcinoma (5).
Chronic Liver Disease and the Ductular Reaction
CLD induces molecular and cellular processes, which are initially reparative but become detrimental in the prolonged setting. Damaged liver epithelial cells release pro-inflammatory signalling molecules, which recruit immune cells to the site of injury, induce collagen deposition or fibrosis and activate liver progenitor cells (LPCs) as part of the so-called ‘ductular reactions’ to restore lost liver tissue. The term ductular reaction describes the diverse histological phenomena occurring in response to chronic hepatic injury and encompasses the epithelial component as well as inflammatory and fibrogenic changes (6). Ductular reactions are observed in all forms of CLD with hepatocyte injury and replicative arrest. However, depending on the underlying aetiology, they display diverse morphologies, ranging from well-formed ductules to irregular strings of cells without obvious lumina (7). Irrespective of the injury stimulus, ductular reactions, activated biliary epithelial cells and LPCs are generally closely associated with inflammatory cell populations and fibrosis-driving, activated hepatic stellate cells, forming a very dynamic injury and regeneration niche (Figure 1). Although significant differences in injury and repair dynamics can be observed in different forms of CLD (8), epithelial, inflammatory and fibrogenic cells principally orchestrate liver regeneration versus disease progression through chemokine and cytokine crosstalk in all clinical settings (6, 9–13).
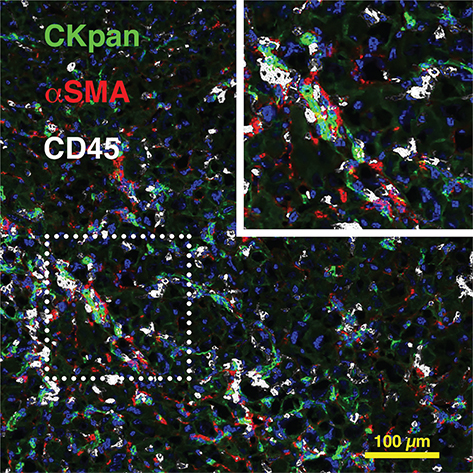
Figure 1 The injury and regeneration niche during chronic liver injury. Murine chronic liver injury induced by feeding a choline-deficient, ethionine-supplemented diet 15 leads to formation of an injury and regeneration niche, involving CKpan+ ductular cells and LPCs (green), αSMA+ hepatic stellate cells (red) and CD45+ inflammatory cells (white). DAPI was used for nuclear localisation.
Liver Progenitor Cells and Cancer Stem Cells
LPCs are defined as a heterogeneous pool of immature, bipotential hepatic cells with diverse marker expression profiles and the ability to differentiate into either hepatocytes or biliary epithelial cells, depending on the underlying injury stimulus and thus tissue requirements. They are undetectable in healthy liver but upon injury emerge in portal areas near the Canals of Hering. Their origin and liver repopulation capacity have been controversially discussed (14). Studies using the choline-deficient, ethionine-supplemented model of chronic liver injury (15) reported that LPCs expressing osteopontin (16) or Foxl1 (17) contributed to hepatocellular regeneration. In addition, transplantation of clonogenic LPCs into hepatocyte-senescent murine livers, induced through deletion of the E3 ubiquitin ligase Mdm2, resulted in restoration of the hepatic parenchyma through generation of hepatocytic or biliary epithelia (18). The exact underlying mechanisms of LPC-mediated liver regeneration are not always clear; however, hepatocyte senescence seems to be a definite histological requirement (19).
The degree of LPC proliferation directly correlates with the severity of hepatocyte replicative arrest and the inflammatory and fibrogenic responses to CLD (20). Targeting of c-kit+ LPCs through the multikinase inhibitor imatinib mesylate during experimental chronic liver injury resulted in reduced fibrogenesis and carcinogenesis (21). Moreover, the presence of hepatobiliary LPCs, marked by epithelial cell adhesion molecule (EpCAM) and cytokeratin 7 and 19, predicted an increased risk of tumour formation in cirrhotic, hepatitis C virus–infected patients (22). This suggests that some LPCs either indirectly influence tumour development by regulating the fibrogenic potential and chemotaxis of neighbouring hepatic stellate cells (9, 12, 13, 23) or directly as tumour-initiating or cancer stem cells (CSCs).
In general, CSCs are defined as undifferentiated cells that are capable to self-renew, initiate and maintain tumour growth and may be responsible for tumour recurrence after resection. Haraguchi and colleagues first postulated the existence of liver CSCs, based on the finding that the hepatocellular carcinoma cell lines HuH7 and Hep3B contained 0.9–1.8% of side population cells with the ability to efflux the fluorescent nucleic acid-binding dye Hoechst 33342 through high activity of adenosine triphosphate-binding cassette transporters (24). Similar side population cells successfully induced xenograft tumours upon transplantation into immunodeficient NOD/SCID mice, while no tumour formation was observed when non-side population cells were transplanted (25). Subsequently, numerous studies have focussed on the identification of reliable marker expression profiles for liver CSCs. The CD133+ subpopulation of various hepatocellular carcinoma cell lines displayed a more immature, proliferative phenotype with greater colony formation capacity in vitro and upon xenotransplantation a higher tumorigenic potential compared to the CD133− cellular counterpart (26–28). Within the CD133+ population, cells with the expression profile CD133+CD44+ have been described as more tumorigenic and metastatic than CD133+CD44− cells (29, 30). Other studies have suggested the mucin-like cell surface glycoprotein CD24 (31) and the glycosylphosphatidylinositol-anchored glycoprotein CD90 or Thy-1 (32) as liver CSC markers. The transmembrane glycoprotein EpCAM is expressed by normal LPCs and CSCs and regulates cell–cell adhesion, proliferation, migration, differentiation and invasion (33). EpCAM is transcriptionally activated by the Wnt/β-catenin pathway, while inhibition of Wnt/β-catenin signalling was shown to suppress its expression (34). Interestingly, both CD44 and CD24 are direct Wnt target genes, marking this signalling pathway a key player in CSC biology and therefore a potential therapeutic target to prevent or treat hepatocellular carcinoma.
There is strong experimental evidence for Wnt signalling directly regulating the biology of LPCs and CSCs. Using the 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) model of chronic liver injury, Hu et al. demonstrated Wnt/β-catenin signalling activity in proliferating A6+ LPCs and ductular reactions. Primary LPCs showed active, nuclear β-catenin and entered the cell cycle upon Wnt3a stimulation in vitro (35). A constitutively active β-catenin mutant was shown to promote LPC expansion in rodents subjected to the 2-acetylaminofluorene/partial hepatectomy model. In addition, the less differentiated, LPC-like, OV6+ subpopulation of hepatocellular carcinoma cells displayed endogenously active Wnt/β-catenin signalling, coupled with a more aggressive phenotype, as judged by greater tumorigenicity and chemoresistance (36). Boulter and colleagues reported Wnt3a-induced expression of the ubiquitin ligase Numb, which is required to leave the biliary differentiation path, and hepatocyte nuclear factor 4α in the LPC line BMOL (37), together inducing its differentiation towards the hepatocyte lineage (38).
The Wnt/β-Catenin Signalling Pathway
The Wnt signalling pathway is highly conserved and has been associated with embryogenesis, proliferation, differentiation as well as carcinogenesis (39–41). It consists of 19 Wnt ligands, 10 Wnt receptors, referred to as frizzleds (FZD), a family of co-receptors, including low-density lipoprotein receptor-related proteins 5 and 6 (LRP5 and 6), and two branches of the pathway exist (42, 43). The non-canonical pathway comprises the planar cell polarity pathway (PCP) and the Ca2+ pathway. These are β-catenin-independent pathways that play roles in the regulation of the actin cytoskeleton and cytoskeletal rearrangement and will not be discussed further. In contrast, the canonical pathway is β-catenin-dependent and of particular interest therapeutically, as aberrant activation of this pathway has been postulated as a key driver in many malignancies such as prostate, colorectal, ovarian and liver cancer (41).
During the inactive ‘off’ state, there is no Wnt ligand bound to the FZD receptor, which results in the multi-protein destruction complex, consisting of glycogen synthase kinase 3β (GSK3β), casein kinase 1 (CK1), adenomatous polyposis coli (APC) and Axin, to bind β-catenin. The destruction complex then phosphorylates β-catenin in a sequential pattern on residues serine 33 (S33), serine 37 (S37) and threonine (T41). Beta-catenin is then ubiquitinated by the E3-ligase beta-transducin repeat containing protein (βTRCP) and marked for proteasomal degradation, preventing it from translocating to the nucleus. The transcription repressor Groucho remains bound to T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors, inhibiting transcription of target genes such as c-Myc and cyclin D1 (Figure 2) (40). Conversely, in the active ‘on’ state, a Wnt ligand binds to a FZD receptor, activating the protein dishevelled (Dvl), a cytoplasmic phosphoprotein crucial for Wnt signal transduction. Axin is recruited to the plasma membrane, binding to the co-receptor LRP5/6 and inhibiting GSK3β and the destruction complex. This allows unphosphorylated β-catenin to accumulate in the cytoplasm and translocate into the nucleus, where Groucho is displaced and unbound from TCF/LEF transcription factors. In this case, β-catenin is able to bind and activate downstream signalling (Figure 2) (40). It has been estimated that Wnt/β-catenin signalling regulates the expression of more than 80 target genes involved in cell fate determination, development, regeneration, zonation, metabolism, fibrosis and carcinogenesis of the liver (44, 45).
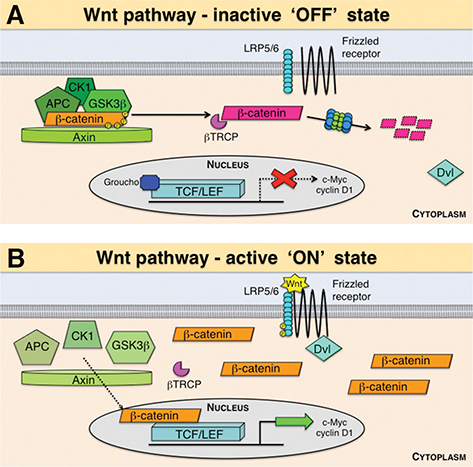
Figure 2 The canonical Wnt/β-catenin pathway. In the absence of a Wnt signal (‘OFF’ state), the destruction complex, consisting of adenomatosis polyposis coli (APC), glycogen synthase kinase 3-β (GSK3β), casein kinase 1 (CK1) and Axin, binds and phosphorylates β-catenin, marking it for ubiquitination by the E3 ubiquitin ligase subunit beta-transducin repeat containing protein (βTRCP) and degradation through the proteasome. In this case, the repressor Groucho remains bound to T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors, inhibiting transcription of target genes such as c-Myc and cyclin D1 (A). When a Wnt protein binds a Frizzled receptor and the low-density lipoprotein receptor-related proteins 5 and 6 (LRP5/6) in the ‘ON’ state, the protein dishevelled (Dvl) activates a cascade, which eventually disrupts the destruction complex, leading to stabilisation, cytoplasmic accumulation and nuclear translocation of β-catenin and ultimately the transcription of target genes (B).
Wnt/β-Catenin Signalling in Liver Metabolism
The liver regulates metabolic homeostasis by controlling glycogen storage, gluconeogenesis, plasma protein synthesis, lipoprotein synthesis and detoxification. To manage fluctuating metabolic demands, hepatic cells constantly alter the expression of respective regulatory pathways. Accordingly, hepatic Wnt signalling activity is modified under different physiological and pathophysiological conditions (45). In adult healthy hepatocytes, β-catenin is ubiquitously expressed, but it is more active in pericentral compared to periportal hepatocytes (44). Expression of β-catenin in periportal regions is inhibited by hepatocyte nuclear factor 4α (46). In contrast, pericentral hepatocytes display basal activation of β-catenin signalling, controlling expression levels of glutamine synthetase, ornithine aminotransferase and the glutamate transporter GLT-1, which together regulate glutamine metabolism (47). This heterogeneous distribution of metabolic function across the lobule reflects hepatic zonation and is necessary to achieve optimal metabolic regulation. Benhamouche and colleagues established that the Wnt/β-catenin pathway is a major control switch pathway for metabolic zonation by demonstrating that blocking of β-catenin in hepatocytes by infection with an adenovirus encoding the Wnt signalling antagonist Dickkopf-1 (Dkk-1) resulted in expansion of the periportal transcriptome and downregulation of perivenous genes. Conversely, constitutive activation of β-catenin through liver-induced disruption of the negative regulator APC reversed this gene expression profile and induced the perivenous gene expression programme (48).
The localisation and signalling activity of β-catenin becomes modified upon liver injury (Figure 3) (44, 49). Debebe and colleagues demonstrated recently that hepatic steatosis experimentally induced by feeding of a high fat diet, deletion of phosphatase and tensin homologue deleted on chromosome 10 (Pten) or transgenic expression of HCV core/NS5A protein, all resulted in macrophage-secreted Wnt activating CD133+CD49f+ tumour-initiating cells. These data strongly suggested a Wnt/β-catenin-mediated link between obesity and cancer (50). In addition, β-catenin was shown to regulate hepatic gluconeogenesis during starvation and insulin-resistant conditions via interaction with the transcription factor forkhead box protein O 1 (FoxO1). This interaction leads to a change in expression of genes encoding the enzymes glucose-6-phosphatase and phosphoenolpyruvate carboxykinase, which then determine the rate of hepatic gluconeogenesis (44, 51). During oxidative stress conditions, β-catenin interacts with FOXO and enhances FOXO transcriptional activity to induce expression of targets for detoxification of reactive oxygen species (52). FOXO factors are sensitive to increased insulin levels, hence the interaction of β-catenin and FOXO is particularly important in diseases associated with insulin resistance, such as non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH) and the metabolic syndrome in general (44). The metabolic syndrome, previously known as the insulin-resistance syndrome, has been defined as a clustering of the risk factors, namely, central obesity, hypertension, hypertriglyceridaemia, hyperglycaemia and low levels of high-density lipoprotein (53). Metabolic syndrome as well as NAFLD are associated with reduced insulin sensitivity and decreased insulin effects on glucose and lipid metabolism (54).
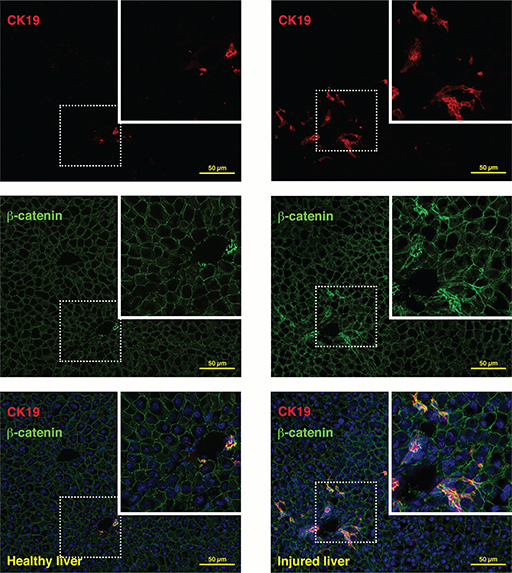
Figure 3 Beta-catenin and CK19 expression in healthy and injured liver. In healthy mouse liver, only ducts stain with an antibody targeting CK19 and show cytoplasmic and nuclear β-catenin expression, while β-catenin is exclusively membrane-bound in periportal hepatocytes (healthy liver, left panel). In injured liver (2-week treatment with a choline-deficient, ethionine-supplemented diet15), the CK19+ compartment expands and demonstrates strong cytoplasmic and nuclear β-catenin, signifying active signalling (injured liver, right panel).
Numerous studies have established a role of the Wnt/β-catenin pathway in the metabolic syndrome since it was demonstrated that Wnt signalling represents a molecular switch to control adipogenesis. Activation of the canonical Wnt/β-catenin pathway through Wnt10b, inhibition of GSK3β or expression of dominant stable β-catenin prevented differentiation of preadipocytes and myoblasts through inhibition of the adipogenic transcription factors CCAAT/enhancer-binding protein α (C/EBPα) and peroxisome proliferator-activated receptor γ (PPARγ) (55, 56). Kennell and MacDougald investigated Xenopus Wnt8 and FZD1 or FZD2 chimeras and established a role for both β-catenin-dependent and β-catenin-independent mechanisms in mesenchymal cell fate and adipogenesis (57). Conversely, in a recent report, inhibition of Wnt signalling through the Wnt-inhibitory molecule sclerostin led to spontaneous adipogenesis of pre-adipocytes and mesenchymal precursors (58), supporting the concept of Wnt signalling controlling the adipogenic switch.
Wnt/β-Catenin Signalling and Cancer
In hepatocellular carcinoma cells, the most frequent mutations occur in TP53, coding for the tumour suppressor p53, and CTNNB1, the β-catenin gene (59). Coste and colleagues suggested that 26% of human and 50% of mouse hepatocellular carcinomas carry activating mutations in CTNNB1 (60). Subsequent studies supported these findings and reported up to 44% of hepatocellular carcinomas with CTNNB1 mutations (61–64). Activation of β-catenin is hypothesised to be due to mutations in exon-3 at the serine and/or threonine sites near the NH2 terminus in CTNNB1, which inhibit phosphorylation-dependent degradation of the β-catenin protein, leading to an aberrant activation of the canonical Wnt/β-catenin pathway (44). Simultaneous mutation of the β-catenin gene and the tumour suppressor H-ras or p21 by an adenovirus-mediated liver-specific Cre-expression system resulted in 100% tumour incidence with short latency of only several weeks (65). Interestingly, mutations in the β-catenin gene in hepatocellular carcinomas induce overexpression of β-catenin targets such as the glutamine synthetase gene GLUL (66, 67), whereas hepatocytes from glutamine synthetase-negative tumours are often H-ras or BRAF mutated and express E-cadherin, reflecting perivenous and periportal profiles, respectively (68). Loss-of-function in APC and Axin is mutually exclusive to CTNNB1 mutations and has been detected in 1–3% and 8–15% of hepatocellular carcinoma cases (for review, see (69)).
In a 2009 study, the Wnt ligands Wnt3, Wnt9a and Wnt10b were shown to be highly expressed in most hepatocellular carcinoma cell lines, irrespective of their differentiation status. Clear profiles were, however, observed with Wnt2b, Wnt4, Wnt5a, Wnt5b and Wnt7b, which were overexpressed in poorly differentiated cell lines, while Wnt8b and Wnt9b were only expressed in well-differentiated cell lines. These data suggested canonical Wnt signalling activity in well-differentiated cells, contributing to tumour initiation and its repression in poorly differentiated cell lines, which the authors hypothesised to regulate tumour progression (70). Other Wnt pathway components associated with hepatocellular carcinoma development include Wnt signalling antagonists such as secreted frizzled-related proteins (SFRPs), Wnt-inhibitory factor (WIF)-1 and Dickkopf (Dkk) proteins. SFRP1 has been suggested as a tumour suppressor gene, since its expression was downregulated due to promoter hypermethylation in 76.1% of hepatocellular carcinoma specimens at the RNA level and in 30% at the protein level (71). In hepatocellular carcinoma cell lines and clinical specimens, WIF-1 expression was equally found to be repressed by promoter hypermethylation, suggesting epigenetic inactivation as the primary cause for WIF-1 loss during hepatocarcinogenesis (72). Inactivity of the negative Wnt regulators Dkk2 and Dkk3 has been reported in human gastrointestinal tumours (73). Fatima and colleagues observed significantly reduced mRNA expression of Dkk4 in almost half of all investigated hepatocellular carcinoma cases. Immunohistochemical data linked decreased Dkk4 expression to accumulation of β-catenin in hepatocellular carcinoma tissue. In addition, the authors showed that Dkk4 overexpression in hepatocellular carcinoma cell lines resulted in reduced cell proliferation, colony formation and cell migration, suggesting a tumour-suppressive role for Dkk4 (74). A recent study demonstrated that hepatocellular carcinoma cells proliferate upon stimulation in high glucose conditions as a result of Dkk4 downregulation, allowing Wnt3a-mediated β-catenin signalling and c-Myc upregulation (75), suggesting the Wnt pathway may be a therapeutic target in insulin-resistant conditions, leading to hepatocellular carcinoma.
Conclusion
Many diverse signalling pathways regulate liver development, homeostasis, regeneration and carcinogenesis. Given the strong evidence for an association of (i) progressive liver disease and LPCs, (ii) CSC-like LPCs and liver tumour formation and (iii) obesity, insulin resistance, hepatic steatosis and hepatocarcinogenesis, and the fact that the Wnt/β-catenin signalling seems to be playing major roles in all these processes, this pathway represents a particularly promising therapeutic target to prevent or treat hepatocellular carcinoma.
Conflict of interest statement
The authors report no conflict of interest with respect to research, authorship and/or publication of this article.
References
- Fernandez-Iglesias A, Gracia-Sancho J. How to face chronic liver disease: The sinusoidal perspective. Front Med (Lausanne). 2017;4:7. http://dx.doi.org/10.3389/fmed.2017.00007
- Williams R, Aspinall R, Bellis M, Camps-Walsh G, Cramp M, Dhawan A, et al. Addressing liver disease in the UK: A blueprint for attaining excellence in health care and reducing premature mortality from lifestyle issues of excess consumption of alcohol, obesity, and viral hepatitis. Lancet. 2014 Nov 29;384(9958):1953–97. http://dx.doi.org/10.1016/S0140-6736(14)61838-9
- Ozakyol A. Global epidemiology of hepatocellular carcinoma (HCC epidemiology). J Gastrointest Cancer. 2017 Jun 19. http://dx.doi.org/10.1007/s12029-017-9959-0
- Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018 Jan 4. http://dx.doi.org/10.1016/S0140-6736(18)30010-2
- Elsegood CL, Tirnitz-Parker JE, Olynyk JK, Yeoh GC. Immune checkpoint inhibition: Prospects for prevention and therapy of hepatocellular carcinoma. Clin Transl Immunology. 2017 Nov;6(11):e161. http://dx.doi.org/10.1038/cti.2017.47
- Williams MJ, Clouston AD, Forbes SJ. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology. 2014 Feb;146(2):349–56. http://dx.doi.org/10.1053/j.gastro.2013.11.034
- Gouw AS, Clouston AD, Theise ND. Ductular reactions in human liver: Diversity at the interface. Hepatology. 2011 Nov;54(5):1853–63. http://dx.doi.org/10.1002/hep.24613
- Kohn-Gaone J, Dwyer BJ, Grzelak CA, Miller G, Shackel NA, Ramm GA, et al. Divergent inflammatory, fibrogenic, and liver progenitor cell dynamics in two common mouse models of chronic liver injury. Am J Pathol. 2016 Jul;186(7):1762–74. http://dx.doi.org/10.1016/j.ajpath.2016.03.005
- Dwyer BJ, Olynyk JK, Ramm GA, Tirnitz-Parker JE. TWEAK and LTbeta signaling during chronic liver disease. Front Immunol. 2014;5:39. http://dx.doi.org/10.3389/fimmu.2014.00039
- Elsegood CL, Chan CW, Degli-Esposti MA, Wikstrom ME, Domenichini A, Lazarus K, et al. Kupffer cell-monocyte communication is essential for initiating murine liver progenitor cell-mediated liver regeneration. Hepatology. 2015 Oct;62(4):1272–84. http://dx.doi.org/10.1002/hep.27977
- Grzelak CA, Martelotto LG, Sigglekow ND, Patkunanathan B, Ajami K, Calabro SR, et al. The intrahepatic signalling niche of hedgehog is defined by primary cilia positive cells during chronic liver injury. J Hepatol. 2014 Jan;60(1):143–51. http://dx.doi.org/10.1016/j.jhep.2013.08.012
- Ruddell RG, Knight B, Tirnitz-Parker JE, Akhurst B, Summerville L, Subramaniam VN, et al. Lymphotoxin-beta receptor signaling regulates hepatic stellate cell function and wound healing in a murine model of chronic liver injury. Hepatology. 2009 Jan;49(1):227–39. http://dx.doi.org/10.1002/hep.22597
- Tirnitz-Parker JE, Olynyk JK, Ramm GA. Role of TWEAK in coregulating liver progenitor cell and fibrogenic responses. Hepatology. 2014 Mar;59(3):1198–201. http://dx.doi.org/10.1002/hep.26701
- Kohn-Gaone J, Gogoi-Tiwari J, Ramm GA, Olynyk JK, Tirnitz-Parker JE. The role of liver progenitor cells during liver regeneration, fibrogenesis, and carcinogenesis. Am J Physiol Gastrointest Liver Physiol. 2016 Feb 1;310(3):G143–54. http://dx.doi.org/10.1152/ajpgi.00215.2015
- Gogoi-Tiwari J, Kohn-Gaone J, Giles C, Schmidt-Arras D, Gratte FD, Elsegood CL, et al. The murine choline-deficient, ethionine-supplemented (CDE) diet model of chronic liver injury. J Vis Exp. 2017 Oct 21(128). http://dx.doi.org/10.3791/56138
- Espanol-Suner R, Carpentier R, Van Hul N, Legry V, Achouri Y, Cordi S, et al. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology. 2012 Dec;143(6):1564–75.e7. http://dx.doi.org/10.1053/j.gastro.2012.08.024
- Shin S, Upadhyay N, Greenbaum LE, Kaestner KH. Ablation of foxl1-cre-labeled hepatic progenitor cells and their descendants impairs recovery of mice from liver injury. Gastroenterology. 2015 Jan;148(1):192–202.e3. http://dx.doi.org/10.1053/j.gastro.2014.09.039
- Lu WY, Bird TG, Boulter L, Tsuchiya A, Cole AM, Hay T, et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat Cell Biol. 2015 Aug;17(8):971–83. http://dx.doi.org/10.1038/ncb3203
- Raven A, Lu WY, Man TY, Ferreira-Gonzalez S, O’Duibhir E, Dwyer BJ, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature. 2017 Jul 20;547(7663):350–4. http://dx.doi.org/10.1038/nature23015
- Prakoso E, Tirnitz-Parker JE, Clouston AD, Kayali Z, Lee A, Gan EK, et al. Analysis of the intrahepatic ductular reaction and progenitor cell responses in hepatitis C virus recurrence after liver transplantation. Liver Transpl. 2014 Dec;20(12):1508–19. http://dx.doi.org/10.1002/lt.24007
- Knight B, Tirnitz-Parker JE, Olynyk JK. C-kit inhibition by imatinib mesylate attenuates progenitor cell expansion and inhibits liver tumor formation in mice. Gastroenterology. 2008 Sep;135(3):969–79, 979.e1. http://dx.doi.org/10.1053/j.gastro.2008.05.077
- Ziol M, Nault JC, Aout M, Barget N, Tepper M, Martin A, et al. Intermediate hepatobiliary cells predict an increased risk of hepatocarcinogenesis in patients with hepatitis C virus-related cirrhosis. Gastroenterology. 2010 Jul;139(1):335–43.e2. http://dx.doi.org/10.1053/j.gastro.2010.04.012
- Pozniak KN, Pearen MA, Pereira TN, Kramer CSM, Kalita-De Croft P, Nawaratna SK, et al. Taurocholate induces biliary differentiation of liver progenitor cells causing hepatic stellate cell chemotaxis in the ductular reaction: Role in pediatric cystic fibrosis liver disease. Am J Pathol. 2017 Dec;187(12):2744–57. http://dx.doi.org/10.1016/j.ajpath.2017.08.024
- Haraguchi N, Utsunomiya T, Inoue H, Tanaka F, Mimori K, Barnard GF, et al. Characterization of a side population of cancer cells from human gastrointestinal system. Stem Cells. 2006 Mar;24(3):506–13. http://dx.doi.org/10.1634/stemcells.2005-0282
- Chiba T, Kita K, Zheng YW, Yokosuka O, Saisho H, Iwama A, et al. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology. 2006 Jul;44(1):240–51. http://dx.doi.org/10.1002/hep.21227
- Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007 Jun;132(7):2542–56. http://dx.doi.org/10.1053/j.gastro.2007.04.025
- Suetsugu A, Nagaki M, Aoki H, Motohashi T, Kunisada T, Moriwaki H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem Biophys Res Commun. 2006 Dec 29;351(4):820–4. http://dx.doi.org/10.1016/j.bbrc.2006.10.128
- Yin S, Li J, Hu C, Chen X, Yao M, Yan M, et al. CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int J Cancer. 2007 Apr 1;120(7):1444–50. http://dx.doi.org/10.1002/ijc.22476
- Piao LS, Hur W, Kim TK, Hong SW, Kim SW, Choi JE, et al. CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett. 2012 Feb 28;315(2):129–37. http://dx.doi.org/10.1016/j.canlet.2011.10.012
- Zhu Z, Hao X, Yan M, Yao M, Ge C, Gu J, et al. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int J Cancer. 2010 May 1;126(9):2067–78. http://dx.doi.org/10.1002/ijc.24868
- Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, Ng IO. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell. 2011 Jul 8;9(1):50–63. http://dx.doi.org/10.1016/j.stem.2011.06.005
- Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008 Feb;13(2):153–66. http://dx.doi.org/10.1016/j.ccr.2008.01.013
- Dolle L, Theise ND, Schmelzer E, Boulter L, Gires O, van Grunsven LA. EpCAM and the biology of hepatic stem/progenitor cells. Am J Physiol Gastrointest Liver Physiol. 2015 Feb 15;308(4):G233–50. http://dx.doi.org/10.1152/ajpgi.00069.2014
- Yamashita T, Budhu A, Forgues M, Wang XW. Activation of hepatic stem cell marker EpCAM by Wnt-beta-catenin signaling in hepatocellular carcinoma. Cancer Res. 2007 Nov 15;67(22):10831–9. http://dx.doi.org/10.1158/0008-5472.CAN-07-0908
- Hu M, Kurobe M, Jeong YJ, Fuerer C, Ghole S, Nusse R, et al. Wnt/beta-catenin signaling in murine hepatic transit amplifying progenitor cells. Gastroenterology. 2007 Nov;133(5):1579–91. http://dx.doi.org/10.1053/j.gastro.2007.08.036
- Yang W, Yan HX, Chen L, Liu Q, He YQ, Yu LX, et al. Wnt/beta-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. 2008 Jun 1;68(11):4287–95. http://dx.doi.org/10.1158/0008-5472.CAN-07-6691
- Tirnitz-Parker JE, Tonkin JN, Knight B, Olynyk JK, Yeoh GC. Isolation, culture and immortalisation of hepatic oval cells from adult mice fed a choline-deficient, ethionine-supplemented diet. Int J Biochem Cell Biol. 2007;39(12):2226–39. http://dx.doi.org/10.1016/j.biocel.2007.06.008
- Boulter L, Govaere O, Bird TG, Radulescu S, Ramachandran P, Pellicoro A, et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med. 2012 Mar 4;18(4):572–9. http://dx.doi.org/10.1038/nm.2667
- Cai C, Zhu X. The Wnt/beta-catenin pathway regulates self-renewal of cancer stem-like cells in human gastric cancer. Mol Med Rep. 2012 May;5(5):1191–6. http://dx.doi.org/10.3892/mmr.2012.802
- Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. http://dx.doi.org/10.1146/annurev.cellbio.20.010403.113126
- Pohl S, Scott R, Arfuso F, Perumal V, Dharmarajan A. Secreted frizzled-related protein 4 and its implications in cancer and apoptosis. Tumour Biol. 2015 Jan;36(1):143–52. http://dx.doi.org/10.1007/s13277-014-2956-z
- Kikuchi A, Yamamoto H, Kishida S. Multiplicity of the interactions of Wnt proteins and their receptors. Cell Signal. 2007 Apr;19(4):659–71. http://dx.doi.org/10.1016/j.cellsig.2006.11.001
- Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Y, et al. LDL-receptor-related proteins in Wnt signal transduction. Nature. 2000 Sep 28;407(6803):530–5. http://dx.doi.org/10.1038/35035117
- Monga SP. Beta-catenin signaling and roles in liver homeostasis, injury, and tumorigenesis. Gastroenterology. 2015 Jun;148(7):1294–310. http://dx.doi.org/10.1053/j.gastro.2015.02.056
- Sethi JK, Vidal-Puig A. Wnt signalling and the control of cellular metabolism. Biochem J. 2010 Mar 15;427(1):1–17. http://dx.doi.org/10.1042/BJ20091866
- Colletti M, Cicchini C, Conigliaro A, Santangelo L, Alonzi T, Pasquini E, et al. Convergence of Wnt signaling on the HNF4alpha-driven transcription in controlling liver zonation. Gastroenterology. 2009 Aug;137(2):660–72. http://dx.doi.org/10.1053/j.gastro.2009.05.038
- Cadoret A, Ovejero C, Terris B, Souil E, Levy L, Lamers WH, et al. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene. 2002 Nov 28;21(54):8293–301. http://dx.doi.org/10.1038/sj.onc.1206118
- Benhamouche S, Decaens T, Godard C, Chambrey R, Rickman DS, Moinard C, et al. Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev Cell. 2006 Jun;10(6):759–70. http://dx.doi.org/10.1016/j.devcel.2006.03.015
- Burke ZD, Tosh D. The Wnt/beta-catenin pathway: Master regulator of liver zonation? Bioessays. 2006 Nov;28(11):1072–7. http://dx.doi.org/10.1002/bies.20485
- Debebe A, Medina V, Chen CY, Mahajan IM, Jia C, Fu D, et al. Wnt/beta-catenin activation and macrophage induction during liver cancer development following steatosis. Oncogene. 2017 Oct 26;36(43):6020–9. http://dx.doi.org/10.1038/onc.2017.207
- Liu H, Fergusson MM, Wu JJ, Rovira, II, Liu J, Gavrilova O, et al. Wnt signaling regulates hepatic metabolism. Sci Signal. 2011 Feb 1;4(158):ra6. http://dx.doi.org/10.1126/scisignal.2001249
- Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005 May 20;308(5725):1181–4. http://dx.doi.org/10.1126/science.1109083
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III). JAMA. 2001 May 16;285(19):2486–97.
- Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, et al. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes. 2001 Aug;50(8):1844–50. http://dx.doi.org/10.2337/diabetes.50.8.1844
- Bennett CN, Ross SE, Longo KA, Bajnok L, Hemati N, Johnson KW, et al. Regulation of Wnt signaling during adipogenesis. J Biol Chem. 2002 Aug 23;277(34):30998–1004. http://dx.doi.org/10.1074/jbc.M204527200
- Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, et al. Inhibition of adipogenesis by Wnt signaling. Science. 2000 Aug 11;289(5481):950–3. http://dx.doi.org/10.1126/science.289.5481.950
- Kennell JA, MacDougald OA. Wnt signaling inhibits adipogenesis through beta-catenin-dependent and -independent mechanisms. J Biol Chem. 2005 Jun 24;280(25):24004–10. http://dx.doi.org/10.1074/jbc.M501080200
- Fairfield H, Falank C, Harris E, Demambro V, McDonald M, Pettitt JA, et al. The skeletal cell-derived molecule sclerostin drives bone marrow adipogenesis. J Cell Physiol. 2018 Feb;233(2):1156–67. http://dx.doi.org/10.1002/jcp.25976
- Nault JC, Zucman-Rossi J. Genetics of hepatobiliary carcinogenesis. Semin Liver Dis. 2011 May;31(2):173–87. http://dx.doi.org/10.1055/s-0031-1276646
- de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998 Jul 21;95(15):8847–51. http://dx.doi.org/10.1073/pnas.95.15.8847
- Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012 May 6;44(6):694–8. http://dx.doi.org/10.1038/ng.2256
- Huang H, Fujii H, Sankila A, Mahler-Araujo BM, Matsuda M, Cathomas G, et al. Beta-catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C virus infection. Am J Pathol. 1999 Dec;155(6):1795–801. http://dx.doi.org/10.1016/S0002-9440(10)65496-X
- Yamamoto Y, Sakamoto M, Fujii G, Tsuiji H, Kenetaka K, Asaka M, et al. Overexpression of orphan G-protein-coupled receptor, Gpr49, in human hepatocellular carcinomas with beta-catenin mutations. Hepatology. 2003 Mar;37(3):528–33. http://dx.doi.org/10.1053/jhep.2003.50029
- Zucman-Rossi J, Benhamouche S, Godard C, Boyault S, Grimber G, Balabaud C, et al. Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene. 2007 Feb 1;26(5):774–80. http://dx.doi.org/10.1038/sj.onc.1209824
- Harada N, Oshima H, Katoh M, Tamai Y, Oshima M, Taketo MM. Hepatocarcinogenesis in mice with beta-catenin and Ha-ras gene mutations. Cancer Res. 2004 Jan 1;64(1):48–54. http://dx.doi.org/10.1158/0008-5472.CAN-03-2123
- Boyault S, Rickman DS, de Reynies A, Balabaud C, Rebouissou S, Jeannot E, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007 Jan;45(1):42–52. http://dx.doi.org/10.1002/hep.21467
- Loeppen S, Schneider D, Gaunitz F, Gebhardt R, Kurek R, Buchmann A, et al. Overexpression of glutamine synthetase is associated with beta-catenin-mutations in mouse liver tumors during promotion of hepatocarcinogenesis by phenobarbital. Cancer Res. 2002 Oct 15;62(20):5685–8.
- Hailfinger S, Jaworski M, Braeuning A, Buchmann A, Schwarz M. Zonal gene expression in murine liver: Lessons from tumors. Hepatology. 2006 Mar;43(3):407–14. http://dx.doi.org/10.1002/hep.21082
- Pez F, Lopez A, Kim M, Wands JR, Caron de Fromentel C, Merle P. Wnt signaling and hepatocarcinogenesis: Molecular targets for the development of innovative anticancer drugs. J Hepatol. 2013 Nov;59(5):1107–17. http://dx.doi.org/10.1016/j.jhep.2013.07.001
- Yuzugullu H, Benhaj K, Ozturk N, Senturk S, Celik E, Toylu A, et al. Canonical Wnt signaling is antagonized by noncanonical Wnt5a in hepatocellular carcinoma cells. Mol Cancer. 2009 Oct 22;8:90. http://dx.doi.org/10.1186/1476-4598-8-90
- Huang J, Zhang YL, Teng XM, Lin Y, Zheng DL, Yang PY, et al. Down-regulation of SFRP1 as a putative tumor suppressor gene can contribute to human hepatocellular carcinoma. BMC Cancer. 2007 Jul 12;7:126. http://dx.doi.org/10.1186/1471-2407-7-126
- Deng Y, Yu B, Cheng Q, Jin J, You H, Ke R, et al. Epigenetic silencing of WIF-1 in hepatocellular carcinomas. J Cancer Res Clin Oncol. 2010 Aug;136(8):1161–7. http://dx.doi.org/10.1007/s00432-010-0763-5
- Sato H, Suzuki H, Toyota M, Nojima M, Maruyama R, Sasaki S, et al. Frequent epigenetic inactivation of DICKKOPF family genes in human gastrointestinal tumors. Carcinogenesis. 2007 Dec;28(12):2459–66. http://dx.doi.org/10.1093/carcin/bgm178
- Fatima S, Lee NP, Tsang FH, Kolligs FT, Ng IO, Poon RT, et al. Dickkopf 4 (DKK4) acts on Wnt/beta-catenin pathway by influencing beta-catenin in hepatocellular carcinoma. Oncogene. 2012 Sep 20;31(38):4233–44. http://dx.doi.org/10.1038/onc.2011.580
- Chouhan S, Singh S, Athavale D, Ramteke P, Pandey V, Joseph J, et al. Glucose induced activation of canonical Wnt signaling pathway in hepatocellular carcinoma is regulated by DKK4. Sci Rep. 2016 Jun 8;6:27558. http://dx.doi.org/10.1038/srep27558