
Download
Review
Impact of obesity and insulin resistance on diabetic kidney disease progression
Ameen Heshmat Ali Rasheedee1, Amit Kumar Singh1,*
1Department of Medical Laboratory Sciences, UIAHS, Chandigarh University, 140301 Mohali, India
Abstract
Diabetic kidney disease is a major microvascular complication of type 2 diabetes mellitus, and as insulin resistance and obesity become more common, the burden of diabetic kidney disease on the healthcare system rises. Epidemiological evidence indicates that, around one-third of people with type 2 diabetes mellitus can develop chronic kidney disease. Obesity and insulin resistance are important factors in the development of kidney injury. Obesity is associated with low-grade inflammation, lipotoxicity, oxidative stress, and adipokine dysregulation, while insulin resistance leads to progressive glomerular injury, altered renal hemodynamics, and hyperinsulinemia. These pathways contribute to albuminuria, hyperfiltration, and eventually a decrease in glomerular filtration rate. Obesity and insulin resistance accelerate the progression of diabetic kidney disease through the induction of hyperfiltration, inflammation, podocyte damage, and fibrotic remodeling. It has been shown that blockade of the rennin angiotensin-aldosterone system (i.e., through angiotensin-converting enzyme inhibitors or angiotensin receptor blockers like losartan) and administration of sodium-glucose cotransporter-2 inhibitors (e.g., empagliflozin) together with weight-loss interventions can slow the progression of diabetic kidney disease, however, a substantial residual risk persists. Existing research gaps indicate limited understanding of thepathophysiological mechanisms, insufficient understanding of renal ectopic fat deposition, and insufficient stratification of patients by insulin resistancephenotypes and obesity. To develop targeted interventions, it is essential to address these gaps through clinical research and comprehensive mechanistic studies. The aim of the study is to synthesize the combined impact of insulin resistance (IR) and obesity on the progression of diabetic kidney disease (DKD). In addition, early diagnosis and targeted treatments are the most important goals for lowering renal and heart complications.
Keywords: Diabetic kidney disease; Type 2 diabetes mellitus; Insulin resistance; Obesity; Glomerular filtration rate; Chronic kidney disease
Submitted: 17 November 2025; Accepted: 06 March 2026; Published: 20 June 2026
Authors for correspondence: Emails: Amit.e15899@cumail.in
How to cite: Ameen Heshmat Ali Rasheedee, Amit Kumar Singh. Impact of obesity and insulin resistance on diabetic kidney disease progression. Journal of Renal and Hepatic Disorders. 2026; 10(1): 24-33. doi: 10.63268/jrenhp.v10i1.255.
DOI: 10.63268/jrenhp.v10i1.255
Copyright: The Author(s). Published by Troika Publisher. License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0
Introduction
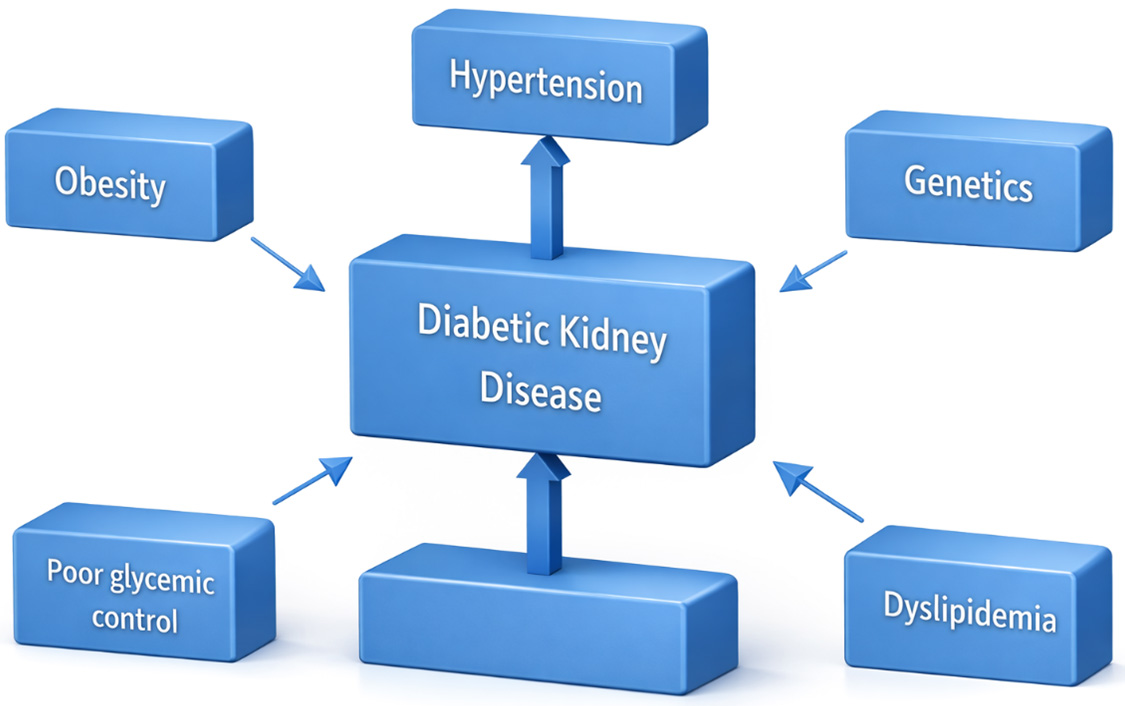
Type 2 diabetes mellitus is a long-term metabolic disease characterized by relative insulin deficiency and insulin resistance, often associated with obesity, dyslipidemia, hypertension, and other features of metabolic disorders [1]. Persistent hyperglycemia in type 2 diabetes mellitus leads to microvascular damage, including in renal glomeruli and tubules, thereby contributing to the development of diabetic kidney disease, a major cause of chronic kidney disease around the globe [2]. Chronic kidney disease in individuals with type 2 diabetes mellitus is highly prevalent: about 27% (95% Confidence interval (CI) 21–33%) of adults globally are affected, according to multiple meta-analyses conducted across multiple countries [3]. Contributing factors to chronic kidney disease among individuals with type 2 diabetes mellitus include older age, longer duration of diabetes, obesity, hypertension, smoking, cardiovascular disease, and possibly a genetic predisposition [3]. Pathophysiologically, hyperglycemia triggers multiple injurious processes in the kidney: glomerular hyperfiltration, increased advanced glycation end products (AGEs), inflammation, oxidative stress, podocyte injury, lipotoxicity, mitochondrial dysfunction, altered tubuloglomerular feedback and tubulo-interstitial fibrosis [2]. Chronic kidney disease in type 2 diabetes mellitus progresses through stages: early kidney damage may be asymptomatic, often first detected via microalbuminuria; over time, albuminuria increases and glomerular filtration rate (GFR) declines, and the risk of end-stage kidney disease rises [1]. Management strategies include blood pressure control (often via agents targeting the renin-angiotensin system), strict glycemic control, use of newer agents such as Glucagon-like peptide-1 (GLP-1) receptor agonists, nonsteroidal mineralocorticoid receptor antagonists, lipid-lowering, and sodium-glucose cotransporter-2 (SGLT2) inhibitors, which have all proven nephroprotective effects in recent trials [4]. Early screening is critical: regular monitoring of estimated GFR (eGFR) and urine albumin excretion allows detection of Chronic Kidney Infection (CKD) at stages when interventions are most effective [5]. Additionally, lifestyle changes (diet, weight management, exercise), smoking cessation, and multidisciplinary care involving patient education are central to slowing CKD progression and improving outcomes [5]. Even with the use of the standard therapies (like Renin-Angiotensin System (RAS) blockade), there remains a residual risk of CKD in individuals with Type 2 Diabetes Mellitus (T2DM), demonstrating the need for new treatments and earlier interventions [6]. Fig. 1 presents a summary of the risk factors related to the progression of diabetic kidney disease.
Figure 1 The risk factors related to the progression of diabetic kidney disease.
This paper is considered a narrative review that describes the existing findings on the impact of obesity and insulin resistance on the progression of diabetic kidney disease. Electronic databases such as PubMed, Google Scholar, and Scopus were used for the literature search. Articles between 2006 and 2025 were identified using keywords such as diabetic kidney disease, obesity, insulin resistance, hyperfiltration, inflammation, and renal outcomes. This review considered several original research articles, clinical trials, observational studies, and meta-analyses. Research focusing on mechanistic pathways, biomarkers, clinical progression, and therapeutic approaches in type 2 diabetes mellitus was prioritized. The selected literature was qualitatively analyzed and interrogated to provide a comprehensive overview of current knowledge and research gaps.
Epidemiology of diabetic kidney disease (DKD)
The burden on the healthcare system of CKD caused by Diabetes Mellitus (DM) has increased markedly over the last decades, with confirmed cases of T2DM-CKD increasing from approximately 0.98 million in 1990 to about 2.50 million in 2019, and prevalent cases increasing in parallel, reaching 129.5 million by 2019 [7]. Mortality and disability due to diabetes-related CKD have also surged: deaths associated with CKD-T2DM reached around 405,990 deaths and DALYs (disability-adjusted life years) about 9.87 million globally by 2019 [7]. There is an increase in the incidence, prevalence, and disease burden of CKD-DM particularly in many countries in Asia [7]. The Simple triage and rapid treatment (START)-India study preliminary findings indicated that about 46% of T2DM patients meet criteria for CKD (urinary albumin-to-creatinine ratio ≥30 mg/g and/or eGFR <60 mL/min/1.73 m2) [8]. Studies indicate that Socially emotional educational knowledge (SEEK)-India reported a CKD prevalence of about 17% in adults, and diabetes was recognized as a major associated factor among the general population of India [9]. Given the large percentage of individuals with diabetes, these data indicate that DKD is a major national and global healthcare burden. Regions with limited medical resources need screening improvement, management strategies, and preventive measures [10].
The primary clinical and metabolic phenotypes that are associated with the progression of diabetic kidney disease including obesity, poor glycemic control, hypertension, dyslipidemia, and genetic risk are shown in this figure. These factors play their roles independently and in combination to increase susceptibility to renal injury in individuals with diabetes.
Pathophysiological basis
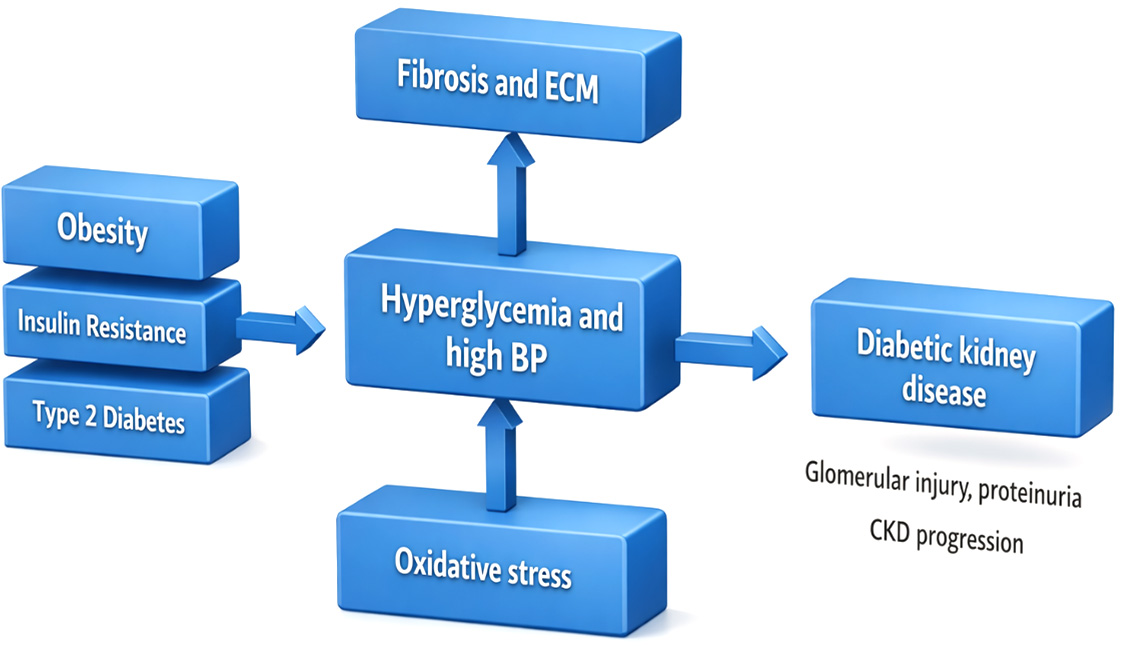
As shown in this Fig. 1, obesity, insulin resistance, and type 2 diabetes are responsible for facilitating diabetic kidney disease via a network of interrelated pathways involving hyperglycemia, elevated blood pressure, oxidative stress, extracellular matrix accumulation, and fibrosis. The outcome of these processes can be glomerular injury, proteinuria, and the emergence of chronic kidney disease.
Role of obesity in metabolic dysfunction
Due to an imbalance between energy intake and expenditure, obesity develops which leads to an increase in adipose tissue mass, which is not a storage site but acts as a hormone-secreting organ that secretes adipokines, free fatty acids, and cytokines that disrupt metabolic homeostasis [11]. Adipose tissue changes to a pro-inflammatory state, especially in visceral adiposity which facilitates systemic metabolic dysfunction and insulin resistance [11]. The metabolic stress is increased by the inflammatory environment across several tissues which disrupt insulin signaling [12]. Endocrine factors are altered by obesity, levels of protective adipokines like adiponectin decrease, whereas leptin resistance, increased resistin, and other adipokine disturbances are involved in reduction of IR [11]. These obesity-related metabolic abnormalities cause metabolic stress, insulin resistance, and chronic inflammation when combined, which increases the development of diabetic kidney disease. Eventually, adaptive hyperinsulinemia is caused by obesity-induced IR, and over time pancreatic β-cell dysfunction ensues, contributing to the progression of overt hyperglycemia and T2DM as metabolic compensation fails [11].
Insulin resistance mechanisms in diabetes
When insulin-target tissues including the liver, adipose tissue, and skeletal muscles are unable to respond properly to normal insulin levels, IR develops, leading to an increase in gluconeogenesis, impaired glucose uptake, and dysregulated lipid metabolism [13]. One primary mechanism includes accumulation of abnormal lipid deposits especially diacylglycerols (DAGs) and ceramides in the liver and muscles, which initiate novel protein kinase C isoforms (e.g., Protein Kinase C (PKC) θ in muscle; PKCϵ in liver) that catalyze phosphorylation of insulin receptor substrate proteins on serine residues, impairing insulin receptor tyrosine kinase signaling [14]. Reduced β-oxidation of fatty acids and increased formation of reactive oxygen species due to mitochondrial dysfunction further facilitate lipotoxicity and oxidative stress, which interfere with insulin signaling cascades and promote inflammation [14]. When nutrient overload occurs (e.g., glucose, excessive free fatty acids), endoplasmic reticulum (ER) stress is induced, triggering the unfolded protein response (UPR) and activating kinases such as Inositol-requiring enzyme1 (IRE1) and Jun N-terminal kinase (JNK), which can transfer a phosphate group to Insulin receptor substrate (IRS) on inhibitory sites, reducing downstream Phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling [14]. Genetic and epigenetic factors (e.g., polymorphisms in IRS, PI3K, insulin receptor genes; alternative splicing; regulation of negative modulators) modulate individual susceptibility to IR [15]. Hyperinsulinemia itself can exacerbate IR by downregulation of insulin receptor expression, induction of receptor internalization, and negative feedback via downstream signaling (e.g., Mammalian Target of Rapamycin (mTOR)/Ribosomal protein S6 kinase (S6K) pathway) that impairs IRS function. Additionally, by creating a chronic state of insulin resistance, the persistence of these defects encourages cellular dysfunction, inflammation, and metabolic stress in insulin-responsive tissues. Together, these mechanisms converge to disrupt the insulin receptor → IRS → PI3K → AKT signaling axis, reduce Glucose transporter type 4 (GLUT4) translocation in muscle and adipose, impair suppression of liver glucose output, and thus chronically elevate blood glucose levels leading to T2DM [14].
Molecular and cellular mechanisms
The major pathophysiological mechanisms linking insulin resistance and obesity to diabetic kidney disease development and progression are seen in Fig. 2.
Figure 2 Pathophysiological mechanisms associating insulin resistance and obesity to diabetic kidney disease development. ECM: Extracellular matrix; BP: Blood pressure; CKD: Chronic kidney disease.
Inflammatory mediators (IL-6, CRP, TNF-α) and adipokines
Adipose tissue secretes adipokines—such as resistin, leptin, and adiponectin—and inflammatory mediators such as Tumor Necrosis Factor Alpha (TNF-α) and Interleukin 6 (IL-6), which play a central role in metabolic dysfunction in obesity [16]. TNF-α released by adipocytes and infiltrating macrophages induces serine phosphorylation of insulin receptor substrate (IRS) proteins, thereby disrupting insulin signaling and contributing to IR [16]. Reduced levels of anti-inflammatory adipokines like adiponectin further change the balance toward pro-inflammatory states, exacerbating IR and renal hemodynamic alterations [16].
Endothelial dysfunction and oxidative stress
Excessive production of reactive oxygen species (ROS) in endothelial cells is caused by oxidative stress driven by metabolic dysregulation and hyperglycemia, overwhelming antioxidant defenses and damaging cellular components [17]. In individuals with diabetes, excessive production of superoxide in endothelial cells activates detrimental pathways, including advanced glycation end products (AGEs), the polyol pathway, the hexosamine pathway, and PKC activation, each contributing to endothelial injury [17].
Initiation of renin-angiotensin-aldosterone system
Systemic and intrarenal Renin-angiotensin-aldosterone system (RAAS) become inappropriately activated in diabetes and obesity, contributing to renal injury and hypertension [18]. The release of renin is stimulated by increased sympathetic tone, hyperglycemia, and signals from the macula densa, resulting in elevated angiotensin II, which produces vasoconstrictive and pro-fibrotic effects [19]. The progression of DKD is promoted by angiotensin II, through stimulation of renal fibrosis and mesangial growth via Transforming growth factor-beta (TGF-β) signaling [18]. The activation of intrarenal RAAS may precede the progression of clinical kidney disease, making RAAS inhibition Angiotensin converting enzyme (ACE) inhibitors or Angiotensin receptor blockers (ARBs) a key factor in preventive therapy in diabetic kidney disease [18].
Podocyte dysfunction and mesangial expansion
The formation of slit diaphragms and maintenance of the filtration barrier are mediated by podocytes, which are highly differentiated glomerular epithelial cells. In DKD, they undergo foot process effacement, loss by apoptosis, hypertrophy, epithelial-mesenchymal transition (EMT), and decreased autophagy, altering structural integrity [20]. Significant causes of podocyte damage that promote apoptosis and structural disruption in DKD include angiotensin II signaling, TGF-β1, Mechanistic/mammalian target of rapamycin complex 1 (mTORC1) activation, and metabolic stress [20]. Experimental studies and clinical research illustrate that podocytes to be one of the initial and most crucial targets of metabolic, inflammatory, and profibrotic attacks in diabetic kidney disease, which resulting in the gradual weakening of glomerular barrier activity. Podocytes are particularly vulnerable to long-term damage in diabetic kidney disease due to these convergent detrimental pathways, which impairs their ability to maintain the integrity of the glomerular filtration barrier. Loss of podocytes is one of the early events in DKD, and correlates with proteinuria and a reduction in GFR. Further, a dysregulation of intracellular calcium (Ca2+) signaling has been found to be a major cause of podocyte impairment in diabetic kidney disease. Metabolic and oxidative stressors disrupt calcium ions homeostasis in the podocyte, triggering cytoskeletal changes, slit diaphragm malfunction, and apoptosis. The interaction of these calcium-dependent processes with inflammatory and profibrotic processes further enhances the glomerular damage and increases proteinuria in diabetic kidney disease [21].
Clinical impact of obesity on DKD progression
Effects on glomerular hyperfiltration
One of the earliest hallmarks of DKD is glomerular hyperfiltration which it can be noticed in individuals with T1DM and T2DM, which occurs before the onset of albuminuria and a gradual decline in renal function [22]. The glomerular capillary pressure is increased by prolonged hyperfiltration, leading to mechanical stress on glomerular structures and promoting mesangial expansion, basement membrane thickening, and podocyte injury [22]. According to several studies, individuals with hyperfiltration have faster GFR decline than those who do not have hyperfiltration, especially when hyperfiltration remains despite treatment [23]. The development from microalbuminuria to macroalbuminuria has been linked to hyperfiltration, indicating its role in disease progression [23]. Molecular profiling of kidneys from hyperfiltering diabetic individuals reveals upregulation of endothelial stress response, TGF-β pathways, and inflammatory signatures, suggesting that hyperfiltration not only induces structural stress, but also activates damaging biochemical cascades [24].
Obesity-related hypertension and proteinuria
Increased body weight canelevate the risk of hypertension through several mechanisms, including sodium retention, overactivation of the sympathetic nervous system, and elevation of RAAS activity, all of which contribute to hypertension in obese people [25]. Obesity also produces structural and functional alterations in the kidney, such as increased GFR hyperfiltration and glomerular capillary wall stress, which predispose to proteinuria [26]. Visceral adipose tissue generates adipokines and cytokines (e.g., leptin, resistin, TNF-α) that promote inflammation, endothelial dysfunction, and increased glomerular permeability [27].
Role of visceral vs. subcutaneous fat
Visceral adipose tissue, which encircles internal structures, is more strongly linked to IR, dyslipidemia, impaired glucose metabolism, metabolic syndrome, and hypertension than subcutaneous adipose tissue (SAT) [28]. SAT, especially in people with lower Visceral adipose tissue (VAT), may have less harmful and potentially protective metabolic effects, partly through better lipid storage and lower secretion of pro-inflammatory cytokines [29]. In longitudinal studies, increases in VAT predict incident metabolic risk factors over time (higher glucose, worse lipid profiles), whereas SAT changes show weaker and less consistent associations after adjusting for VAT [30].
Clinical impact of insulin resistance on DKD progression
Hyperinsulinemia and glomerular injury
The development of DKD and glomerular damage is accelerated by IR-related hyperinsulinemia, which raises intraglomerular pressure and glomerular hyperfiltration [22]. In animal models and early human studies, chronically increased insulin levels provoke glomerular hypertrophy even before the onset of overt hyperglycemia or diabetes, suggesting that hyperinsulinemia alone may initiate structural glomerular changes [31]. Moreover, insulin can stimulate profibrotic pathways, including TGF-β activation, leading to basement membrane thickening and mesangial matrix expansion, thereby accelerating the decline in glomerular filtration rate over time [32].
Insulin resistance and dyslipidemia in DKD
Lipolysis in adipose tissue can be increased by IR in DKD, enhancing free fatty acid (FFA) flux to the liver, which elevates Very Low-Density Lipoprotein (VLDL) production [33]. Some of the characteristics of lipid abnormalities in DKD include elevated triglycerides and increased numbers of small dense Low-Density Lipoprotein (LDL) particles, while High-Density Lipoprotein (HDL) cholesterol tends to be reduced [34]. Lipoprotein lipase (LPL) is suppressed and Apolipoprotein C-III (ApoC-III) is increased by IR, which further impairs triglyceride clearance and worsens dyslipidemia [35]. These lipid disturbances are linked to albuminuria and accelerated decline in eGFR, contributing to the progression of DKD [36].
Link between insulin resistance and accelerated CKD stages
IR is detectable early in CKD, even when glomerular filtration rate is still in the normal or mildly reduced range, and becomes prevalent in most people with severe stages of kidney disease [37]. IR contributes to CKD progression via multiple mechanisms: worsening renal hemodynamics (e.g., increased intraglomerular pressure), sodium retention, and sympathetic nervous system activation [37]. Several cohort studies indicate that higher baseline insulin resistance or hyperinsulinemia is associated with faster progression through CKD stages, greater likelihood of moving from early to moderate and severe CKD [37].
Biomarkers and diagnostic indicators
Key biomarkers for the diagnosis, prognosis, and response to treatment of diabetic kidney disease are seen in Table 1.
Table 1: Most common used and emerging biomarkers in diabetic kidney disease.
| Diagnostic | Therapeutic Response | Prognosis And Follow Up |
| Creatinine | Albuminuria | Creatinine |
| Albuminuria | Glycated haemoglobin | Albuminuria |
| Cystatin | Renal Urine Proteome | Plasma kim-1 |
| Lipocalin | Secretion of Neutrophil | TNF-α |
| Plasma kim-1 | TNFR1 and TNFR2 |
TNF-α: Tumor necrosis factor alpha; TNFR: Tumor necrosis factor receptor.
Role of HbA1c, HOMA-IR, and CRP
Glycated Hemoglobin (HbA1c) values reflect chronic hyperglycemia and are strongly linked to an increased risk of DKD, and HbA1c variability independently correlates with advanced nephropathy in T2DM [38]. HOMA-IR (Homeostatic Model Assessment for Insulin Resistance (HOMA-IR), an abbreviation of homeostasis model assessment of insulin resistance, is increased in individuals with T2DM who develop DKD, and those in the highest tertile of HOMA-IR have significantly greater odds of DKD compared with those in lower tertiles [39]. High-sensitivity CRP (hs-CRP) is correlated with HbA1c and HOMA-IR, and elevated hs-CRP is seen in DKD, reflecting low-grade inflammation as a contributor to renal damage [39]. Thus, together HbA1c (glycemic control), HOMA-IR (insulin resistance), and CRP (inflammation) form an interlinked triad that serves both as a marker of risk and a potential target for intervention to slow DKD progression [39].
Urinary albumin excretion and early renal markers
Albumin-to-creatinine ratio (UACR) or urinary albumin excretion (UAE) are the earliest standardclinical indicator of DKD, correlating with the risk of progression to micro- and macroalbuminuria and declining GFR [40]. However, UAE has limitations: some patients develop renal structural damage or decline in eGFR even while normoalbuminuric, suggesting that albuminuria alone may miss early injury [40]. Consequently, other urinary biomarkers of renal injury are currently under investigation: for example, type IV collagen, urinary N-acetyl-β-D-glucosaminidase (NAG), urinary adiponectin (especially high-molecular-weight forms), and tubular injury markers like retinol-binding protein, α1-microglobulin, and β2-microglobulin [41]. These markers may detect glomerular, podocyte, and tubular damage before overt albuminuria, improving early diagnosis and risk stratification in patients with diabetes [40].
Emerging molecular biomarkers (NGAL, KIM-1, adiponectin, leptin)
Kidney injury molecule-1 (KIM-1) and neutrophil gelatinase-associated lipocalin (NGAL) are elevated in urine in obese children before any decline in GFR or overt proteinuria, indicating early renal tubular injury [42]. In farming communities in Sri Lanka, urinary KIM-1 and NGAL were higher even in apparently healthy individuals in areas with increasing CKD prevalence, suggesting their utility as early detection biomarkers [43]. The adiponectin/leptin ratio and leptin levels have been studied; a higher adiponectin-to-leptin ratio is linked with lower risk of incident CKD, especially in men with lower body mass index [44]. By reflecting early glomerular and tubular injury as well as metabolic and inflammatory abnormalities, these novel biomarkers support their significance in predicting the evolution of diabetic kidney disease and offer insight into disease activity before overt functional deterioration. A recent review of emerging biomarkers in DKD highlights NGAL, KIM-1, adiponectin, and leptin among markers that may improve early risk stratification and potentially guide therapy [45].
Epidemiological and clinical evidence
Population studies linking obesity and DKD
A large cross-sectional study of 4079 Chinese T2DM patients found that both higher Body mass index (BMI) (general obesity) and waist-to-hip ratio (abdominal obesity) were each associated with a higher prevalence of DKD, and those with both general, abdominal obesity and multiple metabolic abnormalities carried the greatest risk of DKD [46]. A British cohort of approximately 4.4 million adults who were metabolically healthy but obese still showed increased risk of incident CKD in comparison to normal-weight healthy individuals (Hazard Ratio (HR) ~1.30 for overweight; ~1.66) for metabolically healthy obesity [47]. A study using Mendelian randomization in T1DM (n ≈ 6049) demonstrated that genetic predisposition to higher BMI causally increases the risk of DKD, macroalbuminuria, and End-stage renal disease (ESRD) [48].
Longitudinal studies on insulin resistance and kidney decline
A 12-year prospective cohort (n = 5347) found that individuals with elevated HOMA-IR trajectory compared with a stable pattern had more than double the risk of adverse renal outcomes (eGFR <60 or new proteinuria), even after adjusting for baseline HOMA-IR and clinical covariates [49]. In the Tehran lipid and glucose study, over 18 years’ follow-up, individuals in the elevated HOMA-IR trajectory group had a significantly higher incidence of CKD (HR ~1.72 in men, ~1.37 in women) compared with stable trajectories [50]. A three-year longitudinal study among Chinese adults without CKD at baseline showed that higher homeostasis model assessment of IR was associated with faster progression to mildly reduced eGFR and CKD; both IR and metabolic syndrome accelerated CKD onset [51]. In elderly Asians (≥65 years), IR and metabolic syndrome were linked with prevalent CKD and more rapid eGFR decline over ~3 years; each unit increase in HOMA-IR raised the odds of CKD and proteinuria. Although the prevalent finding in epidemiological studies indicates clearly that the metabolic syndrome and insulin resistance are crucial determinants associated with chronic kidney disease, the degree of their association with it differs among populations, age groups, and comorbidities, especially in older adults [52].
Therapeutic and preventive approaches
Weight reduction strategies (dietary, surgical, pharmacological)
In patients with CKD, non-surgical interventions such as dietary modifications, exercise, and anti-obesity medications have been shown to reduce BMI by approximately 3.7 kg/m2, and significantly decrease proteinuria (weighted mean difference of approximately 1.31 g/24 h) without accelerating GFR decline [53]. Sustained weight loss with strict metabolic therapies is associated with improvements in renal hemodynamics, reductions in albuminuria, and a reduction in long-term kidney and cardiovascular risk in obese individuals with type 2 diabetes. Bariatric/metabolic surgery (e.g., Roux-en-Y gastric bypass) in obese T2DM patients lowers the incidence of diabetic nephropathy, improves albuminuria, and slows decline of renal function across a range of baseline kidney function levels [54].
Role of SGLT2 inhibitors and RAAS blockers
SGLT2 inhibitors reduce glucose reabsorption in the proximal tubule, increase natriuresis and glucosuria, thereby reducing hyperfiltration, lowering intraglomerular pressure, and improving renal outcomes in DKD [55]. The combination of SGLT2 inhibitors with RAAS blockade offers additive renoprotective benefit: simultaneous suppression of hemodynamic stress, inflammation, and fibrosis appears greater than either agent alone [56]. By restoring tubuloglomerular feedback and demonstrating anti-inflammatory and antifibrotic effects, SGLT2 inhibitors directly target significant pathophysiological pathways that propel the development of diabetic kidney disease, especially in insulin-resistant and obese phenotypes. Therapeutic trials such as Empagliflozin (EMPA-KIDNEY), Dapagliflozin and Prevention of Adverse Outcomes in Chronic Kidney Disease (DAPA-CKD), and Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation (CREDENCE) have shown that SGLT2 inhibitors plus background RAAS inhibitor therapy reduce the risk of kidney endpoints (e.g., sustained GFR decline, ESRD), cardiovascular death or mortality in patients with DKD [57].
Lifestyle modification and exercise interventions
Lifestyle modification combining diet and exercise in obese CKD patients produced significant weight loss (≈5 kg in 12 weeks), reduced fat mass, IR (assessed via the Matsuda index), adipokine profile (increase adiponectin, decrease leptin), and reduced inflammation markers, though GFR remained stable in short term [58]. Intentional weight loss (IWL) strategies—dietary, surgical, and pharmacological—reviewed in obese people with DKD show slowed progression of kidney disease, improvements in albuminuria, but heterogeneity in outcomes due to differing methods and baseline renal function [59]. Lifestyle changes that specifically target sodium intake are crucial for lowering hemodynamic stress and volume overload in individuals with diabetic kidney disease, which slows the progression of chronic kidney disease. Dietary sodium restriction as part of lifestyle counselling in CKD (many with diabetic nephropathy) attenuated proteinuria, reduced blood pressure, and in one study reduced risk of composite renal outcomes over approximately ~3.5 years [60].
Cost-effectiveness of early intervention
Lifestyle interventions in T2DM patients (versus standard diabetes support and education) have been modelled in Japan and shown to be cost-effective in preventing kidney disease: Incremental Cost-Effectiveness Ratio (ICER) ≈ Japanese Yen (JPY) 1,510,838 (~USD 13,031) per Quality-Adjusted Life Year (QALY) gained, with ≈93.6% probability of being cost-effective at a threshold of JPY 5,000,000 per QALY [61]. In India, population-based screening for microalbuminuria among normotensive T2DM patients aged >40 years was cost-effective: ICERs were ~₹ 24,114 (US$ 308) and ₹ 13,790 (US$ 176) per QALY for two screening strategies, reducing ESRD cases significantly and saving large costs over 10 years [62].
Research gaps and future perspectives
Need for mechanistic human studies
It is now recognized that obesity and insulin resistance play a major role in the development of diabetic kidney disease through interconnected hemodynamic, metabolic, inflammatory, and fibrotic pathways. Even though we now have a much better understanding of these pathways, we still have a long way to go before we can effectively translate mechanistic discoveries into phenotype-specific therapies to stop the progression of renal failure. These gaps need to be filled in order to enhance risk classification and therapeutic targeting in obese individuals with diabetic kidney disease. Human kidney biopsy studies are scarce, especially in early disease, which hampers understanding of which cell types (podocytes, endothelial, mesangial or tubularcells) are first altered in obese individuals [63]. The recent integrative studies of insulin-resistant renal models and the small human sample biopsy datasets suggest cell-type-specific molecular alterations that are typical of diabetic kidney disease; however, the time-dependent dynamics of the disease and its pathophysiology are still limited by the experimental modelsand the cross-sectional analysis of human samples. Moreover, longitudinal mechanistic human studies that track molecular, proteomic, transcriptomic and functional changes over time in individuals with obesity and diabetes are few, limiting understanding of causal ordering and potential reversibility. Biomarker studies (e.g., NGAL, KIM-1) show promise, but their mechanistic underpinnings in obese DKD and whether they reflect upstream processes versus downstream damage require clarification. Ultimately, human mechanistic studies are necessary to bridge from bench to bedside: to validate therapeutic targets, identify early disease signatures, and personalize interventions based on mechanisms in obese DKD [64].
Role of genetics and epigenetics in DKD progression
Genetic susceptibility plays a substantial role in DKD: the disease shows familial clustering and heritability in both T1DM and T2DM, suggesting that DNA sequence variation underlies differential risk of developing DKD among individuals exposed to similar metabolic insults. Genome-wide association studies (GWAS) in DKD have detected many genes (e.g., Uromodulin, Engulfment and Cell Motility Protein 1, Carnosine Dipeptidase 1, Apolipoprotein L1, AF4/FMR2 Family Member 3, Advanced Glycosylation End-product Specific Receptor (UMOD, ELMO1, CNDP1, APOL1, AFF3, AGER) whose polymorphisms are associated with susceptibility or resistance to DKD or progression to ESRD. Despite these associations, the effect sizes of individual variants are small, and together they explain only part of the inter-individual variability in DKD risk or progression. Epigenetic modifications such as methylation changes, altered expression of non-coding RNAs, and histone modifications (miRNAs, lncRNAs) mediate how environmental or metabolic exposures (e.g., hyperglycemia, oxidative stress) influence gene expression relevant to DKD progression. For example, epigenome-wide studies (EWAS) have revealed differentially methylated Cytosine-phosphate-Guanine (CpG) sites in kidney-related genes (e.g., Solute Carrier Family 22 Member 12, Transient Receptor Potential Cation Channel Subfamily M Member 6 (SLC22A12, TRPM6) in DKD patients versus controls [65]. Histone methylation modifications (on Histone 3 Lysine 4, Histone 3 Lysine 9, Histone 3 Lysine 27 (H3K4, H3K9, H3K27) have been shown in DKD to contribute to chromatin structural changes that alter transcription of genes driving inflammation, fibrosis, and extracellular matrix deposition [66]. Moreover, the concept of “metabolic memory” depends heavily on epigenetic processes: prior hyperglycemic exposure leaves lasting epigenetic marks that promote DKD progression even after glycemic control is improved [67].
Precision medicine approaches for obesity and insulin resistance in DKD
Precision medicine in T2DM has begun to stratify patients by IR, obesity, and triglyceride levels to predict differential drug responses—e.g., individuals with obesity and high triglycerides show reduced HbA1c response to Dipeptidyl Peptidase-4 (DPP-4) inhibitors compared to non-obese individuals, which may influence the choice of therapy in DKD settings [68]. Recent meta-regression analyses of Randomized Controlled Trials (RCTs) indicate that weight-loss therapies show variable effectiveness depending on patient phenotypes (baseline insulin resistance, obesity degree, metabolic health), suggesting that selecting dietary, pharmacologic, or surgical weight-loss options based on individual traits could enhance outcomes [69]. Genetics is increasingly recognized: obesity genetic variants, polygenic risk scores, and gene-environment interactions might help predict who is more likely to develop IR, renal damage, or respond to weight-loss interventions [70].
Future perspectives
There is a critical need to improve our understanding of DKD progression due to the increasedco-occurrence of obesity and T2DM [25]. Future studies should apply advanced imaging tools such as Computed Tomography (CT) and Magnetic Resonance Imaging (MRI) to quantify perirenal and renal fat deposition and link these findings to renal histopathology. The progression of diabetic kidney disease can be delayed by sodium-glucose cotransporter-2 (SGLT-2) inhibitors through regulation of inflammatory and fibrotic pathways and reduction of intraglomerular pressure. These outcomes are relevant in obese and insulin-resistant patients and, as part of the renoprotective management, support earlier identification of high-risk individuals and timely initiation of SGLT-2 inhibitor therapy [57]. Prospective studies monitoring changes in IR, inflammatory markers, and adiposity are important to clarify the interactive relationship with eGFR decline and albuminuria. In conclusion, to reducing the future burden of DKD affected by obesity and IR, precise therapeutic interventions and bridging mechanistic insights are required [71].
Conclusion
In the development of diabetic kidney disease, obesity and insulin resistance are recognized as major etiological factors, but they are also important drivers of accelerated disease progression. Specifically, the metabolic disturbances of adiposity, insulin resistance, and ectopic lipid deposition promote sustained glomerular hyperfiltration, inflammation, oxidative stress, and fibrotic remodeling, which accelerate structural and functional deterioration of the renal parenchyma. In addition, podocyte injury is a critical downstream event that connects these metabolic and hemodynamic abnormalities to the development of albuminuria and progressive decline in glomerular filtration rate. Importantly, these pathogenic mechanisms often persist even with an adequate level of glycaemic control, and highlighting the need to address non-glycaemic pathways in individuals with type 2 diabetes mellitus and individuals with obesity and insulin resistance. Clinically, obesity-and insulin resistance-related phenotypes need to be identified at an early stage to improve the risk stratification process and to initiate therapeutic interventions as soon as possible. Periodic testing of metabolic biomarkers, insulin resistance indices, and emerging kidney biomarkers could assist in determining patients at high risk of progressive disease. Therapeutic strategies include weight-loss, lifestyle modification, renin-angiotensin-aldosterone system blockade, and sodium-glucose cotransporter-2 blockade have shown specific effectiveness in this cohort, as they simultaneously address the hemodynamic and metabolic predictors of renal injury. Longitudinal human research should be prioritized to clarify the causal relationships underlying the interplay between obesity and insulin resistance in the progression of diabetic kidney disease. Emphasis should be put on cell-type-specific mechanisms, renal ectopic fat deposition, and molecular biomarker integration with clinical endpoints. Finally, the mechanisms-based design of a phenotype-specific therapy holds significant promise for personalized, patient-centered disease management as well as in enhancing renal outcomes in obese individuals with diabetic kidney disease.
Availability of data and materials
Not applicable: This review did not generate or analyze any novel data. All information addressed is obtained as a result of published literature referred to in the article in the past.
Author contributions
AHAR—conceived and designed the review and developed the literature search, data extraction, and analysis. AKS—offered critical advice and helped with literature analysis as well as interpretation of results; helped in editorial modifications, read and endorsed the final manuscript. Both authors initially wrote and edited the manuscript. Both authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Acknowledgment
We would like to acknowledge the department of Medical Laboratory Sciences, UIAHS, Chandigarh University for providing the required facilities.
Funding
This research received no external funding.
Conflict of interest
The authors declare no conflict of interest.
REFERENCES
1. Nordheim E, Geir Jenssen T. Chronic kidney disease in patients with diabetes mellitus. Endocrine Connections. 2021; 10: R151–R159.
2. Francis A, Harhay MN, Ong ACM, Tummalapalli SL, Ortiz A, Fogo AB, et al. Chronic kidney disease and the global public health agenda: an international consensus. Nature Reviews Nephrology. 2024; 20: 473–485.
3. Fenta ET, Eshetu HB, Kebede N, Bogale EK, Zewdie A, Kassie TD, et al. Prevalence and predictors of chronic kidney disease among type 2 diabetic patients worldwide, systematic review and meta-analysis. Diabetology and Metabolic Syndrome. 2023; 15: 245.
4. Dasgupta I, Zac-Varghese S, Chaudhry K, McCafferty K, Winocour P, Chowdhury TA, et al. Current management of chronic kidney disease in type-2 diabetes—a tiered approach: an overview of the joint Association of British Clinical Diabetologists and UK Kidney Association (ABCD-UKKA) guidelines. Diabetic Medicine. 2025; 42: e15450.
5. Jha R, Lopez-Trevino S, Kankanamalage HR, Jha JC. Diabetes and renal complications: an overview on pathophysiology, biomarkers and therapeutic interventions. Biomedicines. 2024; 12: 1098.
6. Chaudhry K, Karalliedde J. Chronic kidney disease in type 2 diabetes: the size of the problem, addressing residual renal risk and what we have learned from the CREDENCE trial. Diabetes, Obesity & Metabolism. 2024; 26: 25–34.
7. Deng Y, Li N, Wu Y, Wang M, Yang S, Zheng Y, et al. Global, regional, and national burden of diabetes-related chronic kidney disease from 1990 to 2019. Frontiers in Endocrinology. 2021; 12: 672350.
8. Prasannakumar M, Rajput R, Seshadri K, Talwalkar P, Agarwal P, Gokulnath G, et al. An observational, cross-sectional study to assess the prevalence of chronic kidney disease in type 2 diabetes patients in India (START-India). Indian Journal of Endocrinology and Metabolism. 2015; 19: 520–523.
9. Singh AK, Farag YM, Mittal BV, Subramanian KK, Reddy SR, Acharya VN, et al. Epidemiology and risk factors of chronic kidney disease in India—results from the SEEK (Screening and Early Evaluation of Kidney Disease) study. BMC Nephrology. 2013; 14: 114.
10. Ma X, Liu R, Xi X, Zhuo H, Gu Y. Global burden of chronic kidney disease due to diabetes mellitus, 1990–2021, and projections to 2050. Frontiers in Endocrinology. 2025; 16: 1513008.
11. Jung UJ, Choi MS. Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. International Journal of Molecular Sciences. 2014; 15: 6184–6223.
12. Martyn JA, Kaneki M, Yasuhara S. Obesity-induced insulin resistance and hyperglycemia: etiologic factors and molecular mechanisms. Anesthesiology. 2008; 109: 137–148.
13. Li M, Chi X, Wang Y, Setrerrahmane S, Xie W, Xu H. Trends in insulin resistance: insights into mechanisms and therapeutic strategy. Signal Transduction and Targeted Therapy. 2022; 7: 216.
14. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiological Reviews. 2018; 98: 2133–2223.
15. Pei J, Wang B, Wang D. Current studies on molecular mechanisms of insulin resistance. Journal of Diabetes Research. 2022; 2022: 1863429.
16. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nature Reviews Immunology. 2011; 11: 85–97.
17. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circulation Research. 2010; 107: 1058–1070.
18. Siragy HM, Carey RM. Role of the intrarenal renin-angiotensin-aldosterone system in chronic kidney disease. American Journal of Nephrology. 2010; 31: 541–550.
19. Peti-Peterdi J, Kang JJ, Toma I. Activation of the renal renin-angiotensin system in diabetes—new concepts. Nephrology Dialysis Transplantation. 2008; 23: 3047–3049.
20. Hu S, Hang X, Wei Y, Wang H, Zhang L, Zhao L. Crosstalk among podocytes, glomerular endothelial cells and mesangial cells in diabetic kidney disease: an updated review. Cell Communication and Signaling. 2024; 22: 136.
21. Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. Journal of Clinical Investigation. 2014; 124: 2333–2340.
22. Tonneijck L, Muskiet MH, Smits MM, van Bommel EJ, Heerspink HJ, van Raalte DH, et al. Glomerular hyperfiltration in diabetes: mechanisms, clinical significance, and treatment. Journal of the American Society of Nephrology. 2017; 28: 1023–1039.
23. Ruggenenti P, Porrini EL, Gaspari F, Motterlini N, Cannata A, Carrara F, et al.; GFR Study Investigators. Glomerular hyperfiltration and renal disease progression in type 2 diabetes. Diabetes Care. 2012; 35: 2061–2068.
24. Stefansson VTN, Nair V, Melsom T, Looker HC, Mariani LH, Fermin D, et al. Molecular programs associated with glomerular hyperfiltration in early diabetic kidney disease. Kidney International. 2022; 102: 1345–1358.
25. Parvanova A, Reseghetti E, Abbate M, Ruggenenti P. Mechanisms and treatment of obesity-related hypertension—part 1: mechanisms. Clinical Kidney Journal. 2023; 17: sfad282.
26. DeMarco VG, Aroor AR, Sowers JR. The pathophysiology of hypertension in patients with obesity. Nature Reviews Endocrinology. 2014; 10: 364–376.
27. Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity, kidney dysfunction and hypertension: mechanistic links. Nature Reviews Nephrology. 2019; 15: 367–385.
28. Hamdy O, Porramatikul S, Al-Ozairi E. Metabolic obesity: the paradox between visceral and subcutaneous fat. Current Diabetes Reviews. 2006; 2: 367–373.
29. Porter SA, Massaro JM, Hoffmann U, Vasan RS, O’Donnel CJ, Fox CS. Abdominal subcutaneous adipose tissue: a protective fat depot? Diabetes Care. 2009; 32: 1068–1075.
30. Abraham TM, Pedley A, Massaro JM, Hoffmann U, Fox CS. Association between visceral and subcutaneous adipose depots and incident cardiovascular disease risk factors. Circulation. 2015; 132: 1639–1647.
31. Zhang Y, Yang S, Cui X, Yang J, Zheng M, Jia J, et al. Hyperinsulinemia can cause kidney disease in the IGT stage of OLETF rats via the INS/IRS-1/PI3-K/Akt signaling pathway. Journal of Diabetes Research. 2019; 2019: 4709715.
32. Sasson AN, Cherney DZ. Renal hyperfiltration related to diabetes mellitus and obesity in human disease. World Journal of Diabetes. 2012; 3: 1–6.
33. Su W, Cao R, He YC, Guan YF, Ruan XZ. Crosstalk of hyperglycemia and dyslipidemia in diabetic kidney disease. Kidney Diseases. 2017; 3: 171–180.
34. Tu QM, Jin HM, Yang XH. Lipid abnormality in diabetic kidney disease and potential treatment advancements. Frontiers in Endocrinology. 2025; 16: 1503711.
35. Kawanami D, Matoba K, Utsunomiya K. Dyslipidemia in diabetic nephropathy. Renal Replacement Therapy. 2016; 2: 16.
36. Ozder A. Lipid profile abnormalities seen in T2DM patients in primary healthcare in Turkey: a cross-sectional study. Lipids in Health and Disease. 2014; 13: 183.
37. Spoto B, Pisano A, Zoccali C. Insulin resistance in chronic kidney disease: a systematic review. American Journal of Physiology-Renal Physiology. 2016; 311: F1087–F1108.
38. Hung CC, Zhen YY, Niu SW, Lin KD, Lin HY, Lee JJ, et al. Predictive value of HbA1c and metabolic syndrome for renal outcome in non-diabetic CKD stage 1–4 patients. Biomedicines. 2022; 10: 1858.
39. Yan H, Zhou Q, Wang Y, Tu Y, Zhao Y, Yu J, et al. Associations between cardiometabolic indices and the risk of diabetic kidney disease in patients with type 2 diabetes. Cardiovascular Diabetology. 2024; 23: 142.
40. Lee SY, Choi ME. Urinary biomarkers for early diabetic nephropathy: beyond albuminuria. Pediatric Nephrology. 2015; 30: 1063–1075.
41. Kern EF, Erhard P, Sun W, Genuth S, Weiss MF. Early urinary markers of diabetic kidney disease: a nested case-control study from the Diabetes Control and Complications Trial (DCCT). American Journal of Kidney Diseases. 2010; 55: 824–834.
42. Polidori N, Giannini C, Salvatore R, Pelliccia P, Parisi A, Chiarelli F, et al. Role of urinary NGAL and KIM-1 as biomarkers of early kidney injury in obese prepubertal children. Journal of Pediatric Endocrinology and Metabolism. 2020; 33: 1183–1189.
43. De Silva PM, Mohammed Abdul KS, Eakanayake EM, Jayasinghe SS, Jayasumana C, Asanthi HB, et al. Urinary biomarkers KIM-1 and NGAL for detection of chronic kidney disease of uncertain etiology (CKDu) among agricultural communities in Sri Lanka. PLOS Neglected Tropical Diseases. 2016; 10: e0004979.
44. Park HS, Park SH, Seong Y, Kim HJ, Choi HY, Rhee Y, et al. Adiponectin-to-leptin ratio and incident chronic kidney disease: sex and body composition-dependent association. Journal of Cachexia, Sarcopenia and Muscle. 2024; 15: 1298–1308.
45. Shetty S, Suvarna R, Awasthi A, Bhojaraja MV, Pappachan JM. Emerging biomarkers and innovative therapeutic strategies in diabetic kidney disease: a pathway to precision medicine. Diagnostics. 2025; 15: 973.
46. Zhang K, Zhang W, Xia F, Wang N, Lu Y, Sui C, et al. Obesity patterns, metabolic abnormality, and diabetic kidney disease in patients with type 2 diabetes. Diabetes, Metabolic Syndrome and Obesity. 2023; 16: 3999–4011.
47. Wang J, Niratharakumar K, Gokhale K, Tahrani AA, Taverner T, Thomas GN, et al. Obesity without metabolic abnormality and incident CKD: a population-based British cohort study. American Journal of Kidney Diseases. 2022; 79: 24–35.e1.
48. Todd JN, Dahlström EH, Salem RM, Sandholm N, Forsblom C, McKnight AJ, et al.; FinnDiane Study Group. Genetic evidence for a causal role of obesity in diabetic kidney disease. Diabetes. 2015; 64: 4238–4246.
49. Yang S, Kwak S, Song YH, Han SS, Lee HS, Kang S, et al. Association of longitudinal trajectories of insulin resistance with adverse renal outcomes. Diabetes Care. 2022; 45: 1268–1275.
50. Amouzegar A, Honarvar M, Masoumi S, Tohidi M, Mehran L, Azizi F. Sex-specific trajectories of insulin resistance markers and reduced renal function during 18 years of follow-up: TLGS. The Journal of Clinical Endocrinology & Metabolism. 2023; 108: e230–e239.
51. Ma A, Liu F, Wang C, Liang K, Yan F, Hou X, et al. Both insulin resistance and metabolic syndrome accelerate the progression of chronic kidney disease among Chinese adults: results from a 3-year follow-up study. International Urology and Nephrology. 2018; 50: 2239–2244.
52. Cheng HT, Huang JW, Chiang CK, Yen CJ, Hung KY, Wu KD. Metabolic syndrome and insulin resistance as risk factors for development of chronic kidney disease and rapid decline in renal function in elderly. The Journal of Clinical Endocrinology & Metabolism. 2012; 97: 1268–1276.
53. Navaneethan SD, Yehnert H, Moustarah F, Schreiber MJ, Schauer PR, Beddhu S. Weight loss interventions in chronic kidney disease: a systematic review and meta-analysis. Clinical Journal of the American Society of Nephrology. 2009; 4: 1565–1574.
54. Liakopoulos V, Franzén S, Svensson AM, Sattar N, Miftaraj M, Björck S, et al. Renal and cardiovascular outcomes after weight loss from gastric bypass surgery in type 2 diabetes: cardiorenal risk reductions exceed atherosclerotic benefits. Diabetes Care. 2020; 43: 1276–1284.
55. DeFronzo RA, Reeves WB, Awad AS. Pathophysiology of diabetic kidney disease: impact of SGLT2 inhibitors. Nature Reviews Nephrology. 2021; 17: 319–334.
56. Zou H, Zhou B, Xu G. SGLT2 inhibitors: a novel choice for the combination therapy in diabetic kidney disease. Cardiovascular Diabetology. 2017; 16: 65. Erratum in: Cardiovascular Diabetology. 2018; 17: 38.
57. Dai ZC, Chen JX, Zou R, Liang XB, Tang JX, Yao CW. Role and mechanisms of SGLT-2 inhibitors in the treatment of diabetic kidney disease. Frontiers in Immunology. 2023; 14: 1213473.
58. Navaneethan SD, Fealy CE, Scelsi AC, Arrigain S, Malin SK, Kirwan JP. A trial of lifestyle modification on cardiopulmonary, inflammatory, and metabolic effects among obese with chronic kidney disease. American Journal of Nephrology. 2015; 42: 274–281.
59. Holland JA, Martin WP, Docherty NG, le Roux CW. Impact of intentional weight loss on diabetic kidney disease. Diabetes, Obesity & Metabolism. 2019; 21: 2338–2341.
60. Kanauchi N, Saito C, Nagai K, Yamada K, Kai H, Watanabe T, et al. Effective method for life-style modifications focused on dietary sodium intake in chronic kidney disease: sub-analysis of the FROM-J study. BMC Nephrology. 2024; 25: 274.
61. Suzuki Y, Hoshi K, Shiroiwa T, Fukuda T. Cost-effectiveness analysis of lifestyle interventions for preventing kidney disease in patients with type 2 diabetes. Clinical and Experimental Nephrology. 2023; 27: 728–736.
62. Mathan Kumar S, Essakky S, Rajasulochana SR, Kar SS, Sivanatham P, Anandraj J, et al. Cost-effectiveness of population-based screening for microalbuminuria in people with type 2 diabetes mellitus in India. International Journal of Technology Assessment in Health Care. 2023; 39: e66.
63. Yau K, Kuah R, Cherney DZI, Lam TKT. Obesity and the kidney: mechanistic links and therapeutic advances. Nature Reviews Endocrinology. 2024; 20: 321–335.
64. Lay AC, Tran VDT, Nair V, Betin V, Hurcombe JA, Barrington AF, et al.; BEAt-DKD Consortium. Profiling of insulin-resistant kidney models and human biopsies reveals common and cell-type-specific mechanisms underpinning Diabetic Kidney Disease. Nature Communications. 2024; 15: 10018.
65. Gu HF. Genetic and epigenetic studies in diabetic kidney disease. Frontiers in Genetics. 2019; 10: 507.
66. Qu P, Li L, Jin Q, Liu D, Qiao Y, Zhang Y, et al. Histone methylation modification and diabetic kidney disease: potential molecular mechanisms and therapeutic approaches (Review). International Journal of Molecular Medicine. 2024; 54: 104.
67. Kato M, Natarajan R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nature Reviews Nephrology. 2019; 15: 327–345.
68. Galiero R, Caturano A, Vetrano E, Monda M, Marfella R, Sardu C, et al. Precision medicine in type 2 diabetes mellitus: utility and limitations. Diabetes, Metabolic Syndrome and Obesity. 2023; 16: 3669–3689.
69. Vargas KG, Rütten T, Siemes B, Brockmeyer M, Parco C, Hoss A, et al. Assessing the potential for precision medicine in body weight reduction with regard to type 2 diabetes mellitus therapies: a meta-regression analysis of 120 randomized controlled trials. Diabetes, Obesity & Metabolism. 2024; 26: 2139–2146.
70. Szczerbinski L, Florez JC. Precision medicine of obesity as an integral part of type 2 diabetes management—past, present, and future. The Lancet Diabetes & Endocrinology. 2023; 11: 861–878.
71. Malik VS, Willett WC, Hu FB. Global obesity: trends, risk factors and policy implications. Nature Reviews Endocrinology. 2013; 9: 13–27.