
Download
Review
Integrating hepatotoxicity monitoring with pharmacogenetics in HIV and tuberculosis treatment: a narrative review
Yair Lara-Blanco1, José R. Chavez-Méndez2, Genaro Rodríguez-Uribe1,*
1Faculty of Medicine and Psychology, Autonomous University of Baja California, 22424 Tijuana, BC, Mexico
2Faculty of Health Sciences “Valle de las Palmas”, Autonomous University of Baja California, 22260 Tijuana, BC, Mexico
Abstract
Drug-induced hepatotoxicity represents a major clinical challenge in the management of patients with Human Immunodeficiency Virus (HIV), tuberculosis (TB), and HIV–TB coinfection, largely due to prolonged and combined therapeutic regimens that increase exposure to potentially hepatotoxic drugs. Early detection and appropriate classification of liver injury remain essential to prevent severe complications and treatment interruptions. This narrative review aims to examine the integration of hepatotoxicity monitoring with pharmacogenetic determinants in the context of HIV and tuberculosis therapy. Current evidence on the epidemiology, pathophysiological mechanisms, diagnostic approaches, and pharmacogenetic predictors of hepatotoxicity is summarized, with particular emphasis on genetic polymorphisms in drug-metabolizing enzymes such as N-acetyltransferase 2 (NAT2) and cytochrome P450 2B6 (CYP2B6), which influence susceptibility to liver injury associated with isoniazid and efavirenz. The available literature indicates that systematic biochemical monitoring, combined with pharmacogenetic information, may improve risk stratification, facilitate early detection of hepatotoxicity, and support more individualized therapeutic strategies. Integrating clinical assessment with pharmacogenetic data could therefore contribute to optimizing treatment safety and advancing personalized medicine approaches in populations affected by HIV and tuberculosis.
Keywords: Hepatotoxicity; HIV infection; Tuberculosis; Coinfection; Pharmacogenetics
Submitted: 26 January 2026; Accepted: 06 May 2026; Published: 20 June 2026
Authors for correspondence: Emails: genaro.rodriguez@uabc.edu.mx
How to cite: Yair Lara-Blanco, José R. Chavez-Méndez, Genaro Rodríguez-Uribe. Integrating hepatotoxicity monitoring with pharmacogenetics in HIV and tuberculosis treatment: a narrative review. Journal of Renal and Hepatic Disorders. 2026; 10(1): 4-13. doi: 10.63268/jrenhp.v10i1.259.
DOI: 10.63268/jrenhp.v10i1.259
Copyright: The Author(s). Published by Troika Publisher. License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0
Introduction
Drug-induced hepatotoxicity (DIH) constitutes a clinically relevant complication in the treatment of Human Immunodeficiency Virus (HIV) infection and tuberculosis (TB). Both conditions require prolonged and combined therapeutic regimens, which increase exposure to multiple drugs and consequently the risk of liver injury [1, 2]. The liver, as the central organ responsible for the biotransformation of antiretroviral and antituberculosis drugs, is particularly vulnerable to injury. Hepatic damage may manifest across a wide clinical spectrum, ranging from asymptomatic elevations of liver enzymes to severe forms of hepatic failure, complicating timely recognition and appropriate management [3, 4].
In the context of HIV–TB coinfection, this risk is further amplified by polypharmacy, drug–drug interactions, and the presence of comorbidities [5]. In this setting, pharmacogenetics has emerged as a valuable tool to explain interindividual variability in treatment response and susceptibility to liver injury. However, its integration with systematic clinical and biochemical assessment of hepatotoxicity remains limited. This gap underscores the need for approaches that consider both components in a complementary manner [1].
A literature search was conducted in PubMed, Embase, and Google Scholar to identify relevant publications addressing hepatotoxicity, HIV, tuberculosis, and pharmacogenetics [6]. Priority was given to studies published within the past six years in order to capture the most recent advances in pharmacogenetics and drug-induced hepatotoxicity research.
Eligible publications included clinical studies, observational studies, systematic reviews, meta-analyses, and clinical practice guidelines that provided relevant clinical or mechanistic evidence related to hepatotoxicity in HIV and tuberculosis treatment. Studies not directly related to hepatotoxicity, pharmacogenetics, HIV, or tuberculosis, as well as articles lacking clinical relevance, were excluded. Seminal earlier references were included when necessary to provide historical or conceptual context.
The objective of this narrative review is to analyze the relevance of hepatotoxicity assessment in the context of HIV and tuberculosis pharmacogenetics, integrating the pathophysiological, clinical, and genetic aspects of this condition, and to discuss its implications for clinical practice and the development of personalized medicine strategies.
Drug-induced hepatotoxicity
Definition
Is a broad clinical concept that describes the presence of structural or functional liver abnormalities associated with exposure to drugs or other xenobiotics. In clinical practice, this term is used to refer to a continuous spectrum of liver injury, which may range from asymptomatic and transient elevations of aminotransferases to clinically significant hepatic lesions [1].
DIH represents one of the leading causes of abnormal liver function tests and of treatment discontinuation or modification. In many clinical scenarios, particularly in settings characterized by polypharmacy or comorbidities, the identification of drug-induced hepatotoxicity allows a pragmatic approach to liver injury, even when it is not possible to definitively establish a direct causal relationship with a specific drug [1, 2].
Causes
DIH is a multifactorial phenomenon resulting from the interaction between drug-related characteristics, host factors, and environmental conditions [2]. Drug-related determinants include dose, duration of treatment, the formation of reactive metabolites during hepatic biotransformation, and the concomitant administration of multiple medications [7]. As the primary organ responsible for xenobiotic metabolism, the liver is particularly vulnerable to these processes, which explains the high frequency of DIH in prolonged and combined treatment regimens, such as those used in HIV and tuberculosis [3].
Host-dependent factors play a central role in individual susceptibility to hepatotoxicity [4]. Variables such as age, sex, nutritional status, alcohol consumption, and the presence of comorbidities and coinfections have been consistently associated with an increased risk of liver injury [8]. Within this context, genetic factors have gained particular relevance, as polymorphisms in genes encoding drug-metabolizing enzymes may alter the pharmacokinetics of antiretroviral and antituberculosis medications [9], promoting the accumulation of hepatotoxic reactive metabolites and increasing the risk of hepatotoxicity, particularly in populations with a high burden of HIV–TB coinfection.
Epidemiology
The epidemiology of DIH is difficult to estimate accurately due to heterogeneity in diagnostic criteria, reporting systems, and the underdiagnosis of asymptomatic cases [2, 3]. At the population level, the annual incidence has been estimated to range from 2 to 20 cases per 100,000 inhabitants, with significant variation across geographic regions and clinical settings [3]. Prospective studies have shown that the true incidence is considerably higher than that reported by pharmacovigilance systems, particularly among hospitalized patients and those exposed to prolonged or combined treatment regimens [4].
In the specific context of tuberculosis, hepatotoxicity associated with antituberculosis drugs represents one of the most frequent adverse drug reactions worldwide [8]. It has been reported that between 2% and 28% of patients receiving first-line treatment regimens develop some degree of hepatotoxicity, with isoniazid, rifampicin, and pyrazinamide being the most commonly implicated drugs [8]. This wide variability in incidence is attributed to differences in study populations, treatment regimens, nutritional status, the presence of comorbidities, and individual genetic predisposition [10].
Among patients with HIV, the prevalence of hepatotoxicity is higher than in the general population and increases significantly in the presence of tuberculosis coinfection [5, 11]. Observational studies and meta-analyses have reported high prevalences of hepatotoxicity in people living with HIV, particularly during the early phases of antiretroviral or antituberculosis treatment [1, 12]. In HIV–TB coinfection, polypharmacy, drug–drug interactions, and alterations in hepatic metabolism contribute to an increased risk of liver injury, underscoring the importance of close clinical and biochemical monitoring in these patients from the initiation of therapy [5].
Types
DIH is classified, based on the pattern of biochemical abnormalities, into hepatocellular, cholestatic, or mixed types [2, 13]. This classification allows for a standardized clinical approach to patterns of liver damage, facilitating both diagnosis and severity assessment. The hepatocellular pattern is characterized by predominant elevations of aminotransferases, particularly alanine aminotransferase (ALT), and is associated with a higher risk of progression to acute liver failure [13]. In contrast, the cholestatic pattern is defined by predominant elevations of alkaline phosphatase (ALP) and bilirubin levels and typically follows a more prolonged course, albeit with lower mortality. The mixed pattern is characterized by concomitant elevations of aminotransferases and ALP, reflecting combined mechanisms of hepatocellular and cholestatic injury [13].
Objective classification of these patterns is achieved using the R ratio, which relates ALT and ALP levels to their respective upper limits of normality (ULN) [2]. This approach has demonstrated clinical utility in standardizing classification and enabling comparison of results across epidemiological and clinical studies [3].
In patients with HIV and TB infection, hepatotoxicity patterns may vary depending on the implicated drug, the presence of concomitant treatments, and individual host-related factors. Antituberculosis drugs are more frequently associated with hepatocellular and mixed patterns, whereas certain antiretroviral agents may induce variable patterns of liver injury, underscoring the importance of systematic biochemical evaluation for accurate characterization of hepatic damage [12].
Pathophysiological and molecular mechanisms of hepatotoxicity
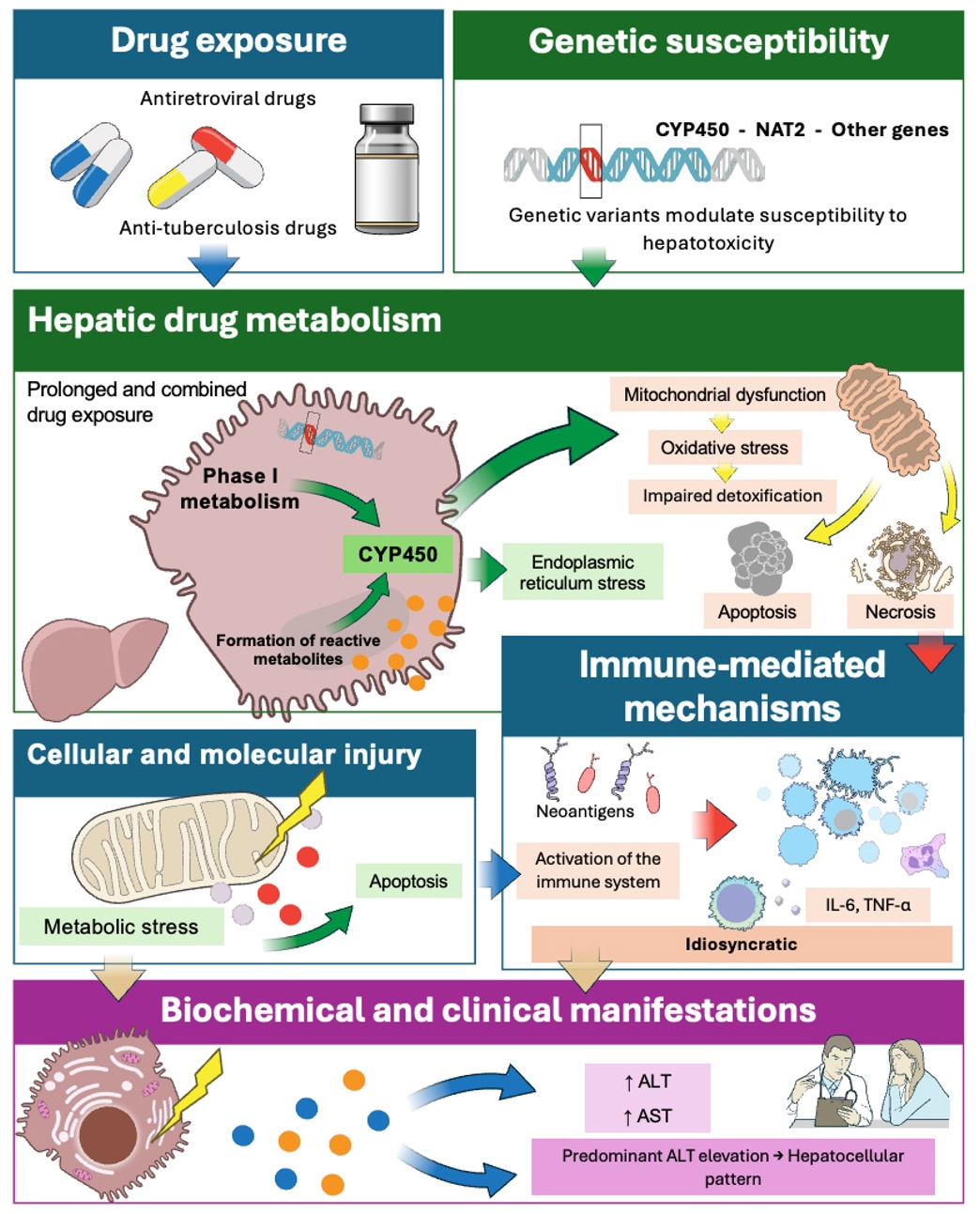
DIH results from a complex interaction between hepatic drug metabolism and host cellular and immunological responses (Fig. 1). Many medications require hepatic biotransformation for elimination through phase I and/or phase II metabolic pathways, processes that may generate reactive metabolites capable of inducing cellular stress, mitochondrial dysfunction, and direct hepatocellular injury [7]. These events can activate cell death pathways, such as apoptosis or necrosis, and trigger an inflammatory response that amplifies liver damage [2].
Figure 1 Pathophysiological and molecular mechanisms of drug-induced hepatotoxicity. Hepatic biotransformation of drugs may generate reactive metabolites that induce mitochondrial dysfunction, oxidative stress, and activation of cell death pathways. In idiosyncratic hepatotoxicity, the formation of neoantigens triggers immune-mediated responses. Genetic factors modulate susceptibility to liver injury. The release of aminotransferases, particularly alanine aminotransferase (ALT), reflects hepatocellular damage. CYP450: Cytochrome P450; NAT2: N-acetyltransferase 2; ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; IL-6: Interleukin-6; TNF-α: Tumor necrosis factor alpha. Source: Own elaboration, with images from bioart.niaid.nih.gov.
In addition to direct toxicity, immune-mediated mechanisms play a key role in DIH. In these cases, drug metabolites may bind to hepatic proteins, forming neoantigens that activate the adaptive immune response. T-lymphocyte activation and the release of proinflammatory cytokines contribute to the progression of liver injury, helping to explain the marked interindividual variability in clinical presentation and disease duration [3].
At the molecular level, genetic factors significantly influence susceptibility to hepatotoxicity. Variants in genes encoding drug-metabolizing enzymes and proteins involved in oxidative stress, endoplasmic reticulum stress responses, and mitochondrial function may modify the liver’s capacity to detoxify drugs [9]. This provides a mechanistic basis for the relevance of pharmacogenetics in identifying individuals at increased risk of hepatotoxicity, particularly in prolonged and combined treatment regimens, such as those used for HIV and tuberculosis.
At the cellular level, aminotransferases, particularly ALT and aspartate aminotransferase (AST), are enzymes predominantly localized in the cytosol and, in the case of AST, also in the mitochondria of hepatocytes. During drug-induced hepatotoxicity, cellular injury mechanisms (including mitochondrial dysfunction, oxidative stress, and activation of cell death pathways) disrupt hepatocellular membrane integrity, facilitating the release of these enzymes into the systemic circulation [7]. Consequently, elevated serum aminotransferase levels serve as an indirect reflection of hepatocellular damage. In particular, predominant ALT elevation is closely associated with the hepatocellular pattern of injury, given its greater hepatic specificity, which explains its widespread use as a key biochemical marker for the identification and characterization of DIH [13].
Distinguishing drug-induced hepatotoxicity from drug-induced liver injury (DILI)
DILI is a specific diagnostic entity that encompasses liver injury attributable to the use of medications administered at therapeutic doses and is considered a diagnosis of exclusion [2]. Its identification requires the systematic exclusion of other causes of liver injury, such as viral hepatitis, autoimmune diseases, metabolic liver disorders, or biliary obstruction. DILI may occur after variable drug exposure and exhibits marked clinical and biochemical heterogeneity, which complicates its timely recognition in clinical practice [3]. In this sense, DILI represents a clinically defined form of drug-induced hepatotoxicity, characterized by the application of more stringent biochemical, temporal, and causality criteria. Therefore, while all cases of DILI involve drug-induced hepatotoxicity, not all manifestations of drug-induced hepatotoxicity meet the clinical and biochemical criteria required to be classified as DILI [1].
From a clinical perspective, DILI is classified as intrinsic or idiosyncratic [2]. The intrinsic form is generally predictable, dose-dependent, and characterized by a short latency period, whereas the idiosyncratic form is unpredictable, not dose-dependent, and exhibits variable latency. The latter is the most common in clinical settings and is influenced by individual factors, including genetic determinants and immune-mediated responses, which explains the marked interindividual variability in its presentation and clinical course [9].
In the context of prolonged and combined treatment regimens, such as those used for HIV and tuberculosis, the identification of cases fulfilling DILI criteria is of particular clinical relevance due to the difficulty in determining the causative drug and the therapeutic implications of discontinuing or modifying essential treatment regimens [5]. In these scenarios, DILI represents a diagnostic and therapeutic challenge that requires systematic and standardized evaluation to minimize the risk of progression to severe liver injury and to optimize treatment continuity.
Evaluation of drug-induced hepatotoxicity
DIH is determined by the presence of liver injury associated with exposure to one or more drugs, identified through biochemical abnormalities, without the need for formal establishment of causality [1]. Unlike DILI, which requires attribution of liver injury to a specific drug, hepatotoxicity may occur across multiple clinical contexts [14, 15]. In clinical studies, hepatotoxicity has historically been defined using operational biochemical criteria. In this regard, the Division of Acquired Immunodeficiency Syndrome (DAIDS) of the National Institutes of Health (NIH) developed standardized criteria for the identification and classification of hepatotoxicity in clinical trials, with the aim of harmonizing the assessment of liver injury severity [16].
Initial evaluation includes measurement of ALT, AST, ALP, gamma-glutamyl transferase (GGT), and bilirubin levels in order to detect and characterize liver injury at an early stage [13, 15]. In populations exposed to polypharmacy, substance use, comorbidities, and coinfections it is often difficult or impossible to establish causality (DILI); therefore, assessment of drug-induced hepatotoxicity becomes essential for the detection, classification, and prevention of liver injury [1].
According to DAIDS criteria, elevations exceeding 1.25 times the ULN constitute the threshold for defining hepatotoxicity, primarily using aminotransferases (particularly ALT) as initial markers of hepatocellular injury [1, 15, 16]. The severity of liver injury can be graded based on the magnitude of enzyme elevation, allowing classification into grade I (mild), grade II (moderate), grade III (severe), and grade IV (potentially life-threatening) toxicity [13, 16]. In addition, the R ratio enables classification of the pattern of liver injury as hepatocellular, cholestatic, or mixed, providing a standardized framework for biochemical interpretation of hepatic damage [13].
To establish a causal relationship between a suspected drug and liver injury, particularly in cases of clinically significant damage, the use of structured methods such as the Roussel Uclaf Causality Assessment Method (RUCAM) is recommended [2]. This scale, specifically developed for the evaluation of DILI, integrates clinical, biochemical, and temporal variables, including the course after drug withdrawal, exclusion of alternative causes, and prior evidence of hepatotoxicity associated with the specific drug [17]. Therefore, RUCAM constitutes a key tool for differentiating nonspecific biochemical elevations from cases that fulfill formal DILI criteria.
Hepatotoxicity in HIV
Hepatotoxicity in patients with HIV is a frequent and multifactorial event, associated both with the use of antiretroviral drugs and with host-related factors. Several antiretroviral agents may induce elevations in liver enzymes through direct or idiosyncratic mechanisms, particularly during the initial phases of treatment or following regimen changes [11]. In addition, factors such as HIV-associated chronic inflammation, the presence of viral hepatitis coinfections, alcohol consumption, and other metabolic comorbidities increase susceptibility to liver injury. In this context, hepatotoxicity may compromise adherence to and continuity of antiretroviral therapy, with clinically relevant implications for patient outcomes [18].
Hepatotoxicity in tuberculosis
In tuberculosis, DIH related to antituberculosis therapy represents one of the most important adverse drug reactions. Isoniazid, rifampicin, and pyrazinamide are the main drugs implicated, and hepatotoxicity typically occurs during the first weeks or months of treatment [19]. The incidence and severity vary widely across populations and are influenced by factors such as age, nutritional status, alcohol consumption, and genetic predisposition. In particular, interindividual variability in isoniazid metabolism partially explains differences in the risk of developing hepatotoxicity during antituberculosis treatment [20].
Hepatotoxicity in HIV-tuberculosis coinfection
Hepatotoxicity in patients with HIV–TB coinfection represents a clinical scenario of particular complexity due to simultaneous exposure to prolonged and potentially hepatotoxic treatment regimens, as well as to clinically relevant drug–drug interactions [5]. Rifampicin, a cornerstone of antituberculosis therapy, acts as a potent inducer of cytochrome P450 enzymes, including cytochrome P450 3A4 (CYP3A4) and cytochrome P450 2B6 (CYP2B6), which can alter the pharmacokinetics of several antiretroviral agents [21].
However, recent studies have demonstrated that enzyme induction does not always translate into increased antiretroviral clearance, as coadministration of isoniazid and genetic variability in drug-metabolizing enzymes may counterbalance these effects. The risk of hepatotoxicity in people living with HIV exposed to preventive or therapeutic tuberculosis regimens is influenced by both the antiretroviral regimen and patient-specific genetic factors. This suggests that drug metabolism in coinfected patients is determined by a complex interaction between pharmacological and genetic determinants, with direct implications for the monitoring and management of liver damage [22].
Pharmacogenetic causes of hepatotoxicity
Drug-induced hepatotoxicity (DIH) exhibits marked interindividual variability that cannot be explained solely by clinical, environmental, or treatment-related factors, highlighting the central role of pharmacogenetics. Genetic variants in enzymes involved in hepatic biotransformation may modify drug metabolism rates, promote the accumulation of reactive metabolites, and impair detoxification mechanisms, thereby increasing susceptibility to hepatotoxicity [1, 23, 24]. In particular, cytochrome P450 (CYP450) enzymes play a fundamental role in phase I metabolism of a wide range of drugs used in clinical practice [25], and polymorphisms in genes encoding these enzymes have been shown to contribute significantly to variability in drug response and toxicity, especially in complex therapeutic regimens such as those used for HIV and tuberculosis [26]. Table 1 (Ref. [23, 26, 27, 28, 29, 30, 31]) summarizes the main pharmacogenetically relevant polymorphisms involved in HIV and tuberculosis treatment and their associated clinical effects.
Table 1: Pharmacogenetically relevant variants associated with variability in drug response and hepatotoxicity risk in tuberculosis and HIV treatment.
| Drug | Gene/Protein | Variant | rsID | Functional phenotype | Main clinical effect | Global allele frequency | Variant type | Change | Position (GRCh38) | |
| Tuberculosis | ||||||||||
| Isoniazid | NAT2 | 5* | rs1801280 | Slow acetylator | ↑ Risk of hepatotoxicity [26, 27] | ~38% | Missense SNP | T > C | chr8: 18400344 | |
| Isoniazid | NAT2 | 6* | rs1799930 | Slow acetylator | ↑ Risk of hepatotoxicity [26, 27] | ~28% | Missense SNP | G > A | chr8: 18400593 | |
| Rifampicin | OATP1B1 (SLCO1B1) | - | rs4149032 | Altered hepatic transport | ↓ Rifampicin exposure [28] | ~37% | Intron variant | C > T | chr12: 21164857 | |
| HIV | ||||||||||
| Efavirenz | CYP2B6 | 6* | rs3745274 | Slow metabolizer | ↑ EFV exposure/hepatic and CNS toxicity [23, 29, 30] | ~26% | Missense SNP | G > T | chr19: 41006936 | |
| Efavirenz/Nevirapine | CYP2B6 | 18* | rs28399499 | Slow metabolizer | ↑ Exposure and toxicity [26, 29] | ~2% | Missense SNP | T > C | chr19: 41012316 | |
| Dolutegravir | UGT1A1 | 28* | rs3064744 | Reduced glucuronidation | ↑ Bilirubin/jaundice [31] | ~30% | Intron variant | TA | chr2: 233760234–233760248 | |
| Dolutegravir | AADAC | - | rs1803155 | Reduced hydrolysis | ↑ Dolutegravir exposure [31] | Variable | Missense SNP | G > A | chr3: 151827813 | |
| ARV/TB (Rifampicin, efavirenz) | PXR (NR1I2) | - | rs2472677 | Altered transcriptional regulation | Alters CYP and transporter expression [28] | ~44% | Intron variant | C > T | chr3: 119799570 | |
NAT2: N-acetyltransferase 2; CYP2B6: Cytochrome P450 2B6; UGT1A1: UDP-glucuronosyltransferase 1A1; AADAC: arylacetamide deacetylase; PXR (NR1I2): pregnane X receptor; SLCO1B1 (OATP1B1): solute carrier organic anion transporter family member 1B1; EFV: efavirenz; CNS: central nervous system; ARV: antiretroviral; TB: tuberculosis; SNP: single nucleotide polymorphism; GRCh38: Genome Reference Consortium Human Build 38; HIV: Human Immunodeficiency Virus.
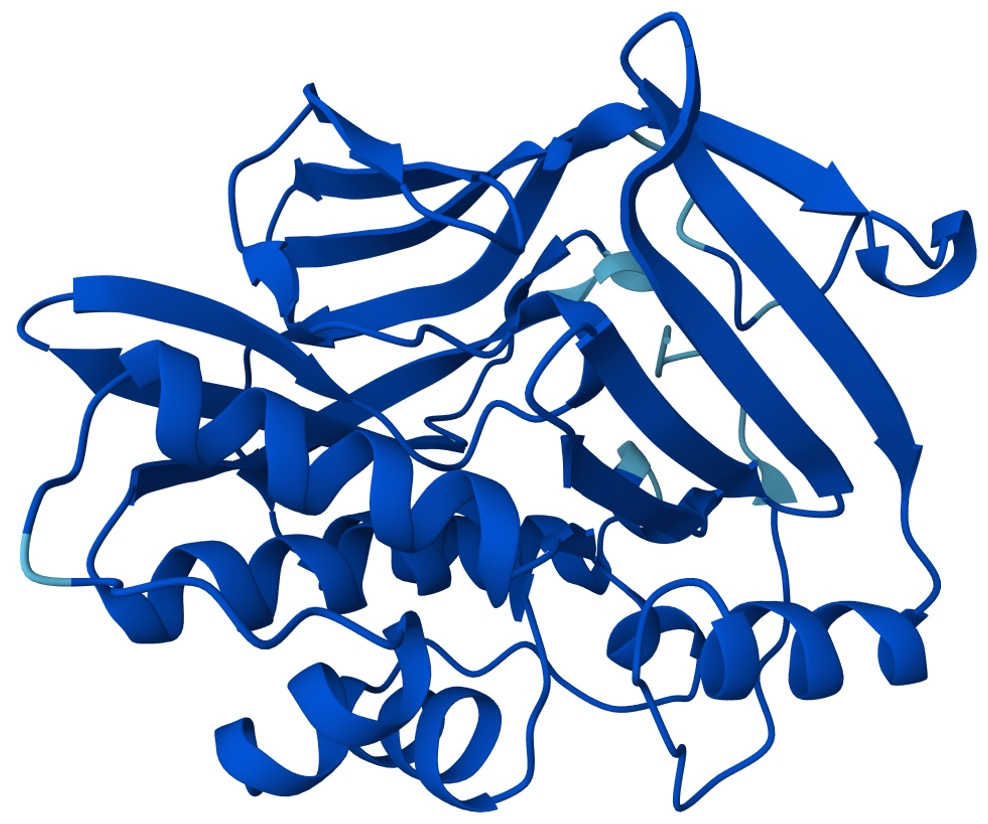
In tuberculosis treatment, N-acetyltransferase 2 (NAT2) represents one of the most relevant pharmacogenetic determinants of hepatotoxicity (Fig. 2) [32]. Polymorphisms in the NAT2 gene define slow, intermediate, or rapid acetylator phenotypes, directly influencing isoniazid metabolism and, consequently, systemic exposure to the drug and its potentially hepatotoxic metabolites [33, 34]. Multiple studies have consistently identified that carriers of NAT2 variants associated with slow acetylation, particularly NAT2*5 (rs1801280, c.341T>C) and NAT2*6 (rs1799930, c.590G>A), have a higher risk of significant ALT elevations and clinically relevant hepatotoxicity during antituberculosis treatment [26, 27]. Accordingly, dose adjustment strategies based on patient-specific factors, including acetylator phenotype, have been proposed [35].
Figure 2 Predicted three-dimensional structure of human N-acetyltransferase 2 (NAT2) obtained from the AlphaFold Protein Structure Database (entry AF-P11245-F1). The structure illustrates the global conformation of the enzyme involved in isoniazid acetylation, whose activity may be altered by genetic polymorphisms associated with an increased risk of hepatotoxicity. Source: Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research. 2022; 50(D1): D439–D444. Data available under a Creative Commons Attribution 4.0 (CC BY 4.0) license.
More recently, clinical evidence has expanded this paradigm by demonstrating that rapid acetylator phenotypes may also have adverse clinical implications [36]. In patients with tuberculosis treated with isoniazid-based regimens, rapid NAT2 acetylators have been associated with lower drug exposure and an increased risk of unfavorable outcomes, including higher one-year mortality, particularly in the context of isoniazid resistance (hazard ratio 1.7–4.6) [36]. Reduced plasma exposure to isoniazid in rapid acetylators, mostly in the presence of drug resistance, may contribute to higher mycobacterial burden and poorer clinical outcomes, including increased mortality.
In patients with HIV, and particularly in those with HIV–tuberculosis coinfection, pharmacogenetics acquires additional complexity due to the interaction of multiple metabolic pathways, enzyme induction and inhibition, and polypharmacy [5, 37, 38]. Among the most extensively studied determinants, cytochrome CYP2B6 stands out as a key regulator of exposure to efavirenz and nevirapine [39, 40]. Polymorphisms such as CYP2B6*6 (rs3745274, c.516G>T), especially when combined with other functional variants, have been consistently associated with elevated plasma efavirenz concentrations and an increased risk of toxicity, including hepatotoxicity [23, 29, 30]. Pharmacokinetic studies in coinfected populations have shown that slow metabolizers exhibit significantly higher efavirenz exposure, even during coadministration with rifampicin and isoniazid, due to inhibition of alternative metabolic pathways such as cytochrome P450 2A6 (CYP2A6) by isoniazid, which may partially counterbalance the enzyme induction exerted by rifampicin [30]. This places pharmacogenetic factors as key determinants of efavirenz exposure and toxicity in specific clinical scenarios [41].
For dolutegravir, plasma concentrations are influenced not only by rifampicin coadministration but also by polymorphisms in enzymes such as UDP-glucuronosyltransferase 1A1 (UGT1A1, rs3064744, 7 TA repeats) and arylacetamide deacetylase (AADAC, rs1803155, c.841G>A), which have been associated with increased drug exposure [31]. In addition, variants in regulatory genes involved in drug metabolism, such as NR1I2 (pregnane X receptor, PXR) rs2472677 (g.24087C>T) and hepatic transporter genes, including SLCO1B1 (solute carrier organic anion transporter family member 1B1) rs4149032 (g.38664C>T), have been shown to independently or jointly modulate exposure to antituberculosis and antiretroviral drugs [28]. In African cohorts with HIV–TB coinfection, PXR polymorphisms have been associated with poorer clinical outcomes, including increased mortality, suggesting that transcriptional regulation of hepatic enzymes and transporters plays a relevant role in interindividual variability beyond classical cytochrome pathways [28].
Taken together, this body of evidence supports that the risk of hepatotoxicity in people living with HIV exposed to isoniazid and antiretroviral therapy is determined by a complex interaction between therapeutic regimens and pharmacogenetic factors, with direct implications for clinical monitoring, risk stratification, and the development of personalized medicine strategies [22].
Pharmacogenetically relevant phenotypes
In HIV–TB coinfection, the pharmacogenetic phenotypes of highest clinical relevance are those that determine increased or decreased exposure to key drugs and, consequently, a higher risk of toxicity, therapeutic failure, or the need for dose adjustments [42, 43]. In tuberculosis, the NAT2 slow acetylator phenotype, derived from allelic combinations of NAT2*5, *6, *7, and *14, is associated with increased isoniazid exposure and a higher incidence of hepatotoxicity, making it a priority phenotype for risk stratification and early biochemical monitoring [27, 33, 43]. Conversely, rapid acetylators represent a subgroup at risk of underexposure and unfavorable clinical outcomes, reinforcing the need to consider pharmacogenetics not only as a tool for toxicity prevention but also for optimization of therapeutic efficacy [36].
Within the antiretroviral component, slow metabolizer phenotypes for efavirenz and nevirapine, primarily associated with variants such as CYP2B6*6 (rs3745274, c.516G>T) and *18 (rs28399499, c.983T>C) are linked to elevated plasma concentrations and an increased likelihood of toxicity, particularly in coinfection settings and during coadministration with antituberculosis drugs [26, 29]. Additional phenotypes derived from variants in regulatory genes (e.g., PXR) or transporter genes (e.g., SLCO1B1) may further modulate exposure to antiretrovirals or rifamycins, generating complex drug–gene–drug interaction scenarios that account for part of the variability observed in clinical practice [28, 44]. Taken together, identification of these pharmacogenetic phenotypes enables conceptualization of clinical heterogeneity in patients with HIV–TB and supports a pharmacovigilance and individualized dosing approach targeted toward subgroups with increased genetic susceptibility [1, 22].
Clinical complications
DIH may lead to a wide spectrum of clinical complications, ranging from transient biochemical abnormalities to severe, life-threatening liver damage, including clinically overt acute hepatitis, prolonged cholestasis, hepatic insufficiency, and, in rare cases, fulminant liver failure [3]. In addition, the occurrence of hepatotoxicity often necessitates the discontinuation or modification of essential therapeutic regimens, which may compromise treatment efficacy, promote disease relapse, and increase the risk of adverse clinical outcomes. These complications are particularly relevant in settings of prolonged or combined therapy, where interruption of the implicated drug represents a significant therapeutic challenge and may be associated with a poorer clinical prognosis [2].
Conclusions
Drug-induced hepatotoxicity remains a clinically relevant complication in the management of patients with HIV, tuberculosis, and HIV–tuberculosis coinfection. Current evidence supports that systematic clinical and biochemical monitoring continues to represent the cornerstone of hepatotoxicity assessment, through periodic evaluation of liver enzymes and the application of standardized hepatotoxicity criteria. Within this framework, pharmacogenetics emerges as a highly valuable complementary tool, enabling the identification of patient subgroups with increased susceptibility to liver injury, particularly in relation to polymorphisms in genes such as NAT2 and CYP2B6. In certain situations, such as resource-limited settings, pharmacogenetic testing may not be routinely available; however, clinical and biochemical monitoring strategies based on liver enzyme assessment remain essential for clinicians in the early detection and management of hepatotoxicity. The integrated evaluation of these elements provides a more precise and preventive approach to the detection and management of hepatotoxicity, with the potential to optimize therapeutic safety and advance personalized medicine strategies in vulnerable populations.
Availability of data and materials
Not applicable.
Author contributions
YLB—conceptualization, manuscript design, and editing. JRCM and GRU—manuscript drafting. All authors contributed to manuscript revision and approved the final version.
Ethics approval and consent to participate
This study is a narrative review and did not involve human participants or identifiable data; therefore, informed consent is not applicable, and ethics committee approval was not required.
Acknowledgment
Not applicable.
Funding
This research received no external funding.
Conflict of interest
The authors declare no conflict of interest.
REFERENCES
1. Cho FN, Achidi EA, Enoh JE, Pallerla SR, Linh LTK, Tong HV, et al. Drug-induced hepatotoxicity and association with slow acetylation variants NAT2*5 and NAT2*6 in Cameroonian patients with tuberculosis and HIV co-infection. BMC Infectious Diseases. 2024; 24: 759.
2. Andrade RJ, Chalasani N, Björnsson ES, Suzuki A, Kullak-Ublick GA, Watkins PB, et al. Drug-induced liver injury. Nature Reviews Disease Primers. 2019; 5: 58.
3. Wang Y, Xie W. Drug-induced liver injury: an overview and update. Gastroenterology & Endoscopy. 2023; 1: 102–109.
4. Suzuki A, Chen M. Epidemiology and risk determinants of drug-induced liver injury: current knowledge and future research needs. Liver International. 2025; 45: e16146.
5. Meintjes G, Maartens G. HIV-associated tuberculosis. The New England Journal of Medicine. 2024; 391: 343–355.
6. Dhillon P. How to write a good scientific review article. The FEBS Journal. 2022; 289: 3592–3602.
7. Skat-Rørdam J, Lykkesfeldt J, Gluud LL, Tveden-Nyborg P. Mechanisms of drug induced liver injury. Cellular and Molecular Life Sciences. 2025; 82: 213.
8. Kumar A, Patel R, Kumar S, Kumar R. Antituberculosis drugs-induced hepatotoxicity: an update. Tropical Gastroenterology. 2024; 45: 125–133.
9. Daly AK. Genetics of drug-induced liver injury: current knowledge and future prospects. Clinical and Translational Science. 2023; 16: 37–42.
10. Nyangwara V, Waja Z, Thelingwani R, Osman R, Pretorius Z, Majoro K, et al. Incidence and associated risk factors of antituberculosis drug-induced liver injury among TB patients. BMC Infectious Diseases. 2025; 25: 1400.
11. Bekker LG, Beyrer C, Mgodi N, Lewin SR, Delany-Moretlwe S, Taiwo B, et al. HIV infection. Nature Reviews Disease Primers. 2023; 9: 42.
12. Mohammed O, Alemayehu E, Bisetegn H, Tilahun M, Gedefie A, Ebrahim E, et al. Prevalence of hepatotoxicity among HIV-infected patients in Ethiopia: a systematic review and meta-analysis. BMC Infectious Diseases. 2022; 22: 826.
13. NIH. LiverTox: clinical and research information on drug-induced liver injury. National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK): Bethesda (MD). 2019.
14. Andrade RJ, Chalasani N, Björnsson ES, Suzuki A, Kullak-Ublick GA, Watkins PB, et al. EASL clinical practice guidelines: drug-induced liver injury. Journal of Hepatology. 2019; 70: 1222–1261.
15. Yimer G, Gry M, Amogne W, Makonnen E, Habtewold A, Petros Z, et al. Evaluation of patterns of liver toxicity in patients on antiretroviral and anti-tuberculosis drugs: a prospective four arm observational study in Ethiopian patients. PLOS ONE. 2014; 9: e94271.
16. Division of AIDS (DAIDS); National Institute of Allergy and Infectious Diseases; National Institutes of Health. Division of AIDS (DAIDS) table for grading the severity of adult and pediatric adverse events. 2017. Available at: https://rsc.niaid.nih.gov/ (Accessed: 17 December 2025).
17. CIOMS/RUCAM scale 2025. Roussel Uclaf Causality Assessment Method (RUCAM). 2025. Available at: https://www.rccc.eu/scores/RUCAM.html (Accessed: 17 December 2025).
18. Gandhi RT, Bedimo R, Hoy JF, Landovitz RJ, Smith DM, Eaton EF, et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2022 recommendations of the International Antiviral Society—USA Panel. JAMA. 2023; 329: 63–84.
19. Trajman A, Campbell JR, Kunor T, Ruslami R, Amanullah F, Behr MA, et al. Tuberculosis. The Lancet. 2025; 405: 850–866.
20. Klindt C, Fuchs A, Behnke K, Dröge C, Eberhardt KA, Orth HC, et al. Genetic and clinical risk factors for anti-tuberculosis drug-induced liver injury: insights from a prospective cohort study in central Ethiopia. Infection. 2025; 53: 2833–2846.
21. HIV.gov Clinical Guidelines. Guidelines for the use of antiretroviral agents in adults and adolescents with HIV. 2025. Available at: https://clinicalinfo.hiv.gov/en/guidelines/hiv-clinical-guidelines-adult-and-adolescent-arv/virologic-failure?view=full (Accessed: 17 December 2025).
22. Montepiedra G, Aaron L, Theron G, McCarthy K, Bradford S, Chipato T, et al. Hepatotoxicity among people living with HIV and receiving isoniazid preventive therapy in pregnancy and postpartum: the role of antiretroviral regimen and pharmacogenetics. Clinical Infectious Diseases. 2026; 82: e156–e164.
23. Nicoletti P, Aithal GP, Bjornsson ES, Andrade RJ, Sawle A, Arrese M, et al. Association of liver injury from specific drugs, or groups of drugs, with polymorphisms in HLA and other genes in a genome-wide association study. Gastroenterology. 2017; 152: 1078–1089.
24. U.S. Food and Drug Administration (FDA). For healthcare professionals — FDA’s examples of drugs that interact with CYP enzymes and transporter systems. 2025. Available at: https://www.fda.gov/drugs/drug-interactions-labeling/healthcare-professionals-fdas-examples-drugs-interact-cyp-enzymes-and-transporter-systems#table%201 (Accessed: 17 December 2025).
25. Zhao M, Ma J, Li M, Zhang Y, Jiang B, Zhao X, et al. Cytochrome P450 enzymes and drug metabolism in humans. International Journal of Molecular Sciences. 2021; 22: 12808.
26. Hardi H, Fitrianti Z, Mahata LE, Louisa M. Pharmacogenetics in tuberculosis–HIV coinfected populations: a systematic review of genetic variants influencing antiretroviral and anti-tuberculosis drug response. Journal of Multidisciplinary Healthcare. 2025; 18: 7203–7218.
27. Gutiérrez-Virgen JE, Piña-Pozas M, Hernández-Tobías EA, Taja-Chayeb L, López-González ML, Meraz-Ríos MA, et al. NAT2 global landscape: genetic diversity and acetylation statuses from a systematic review. PLOS ONE. 2023; 18: e0283726.
28. Calcagno A, Cusato J, Sekaggya-Wiltshire C, von Braun A, Motta I, Turyasingura G, et al. The influence of pharmacogenetic variants in HIV/tuberculosis coinfected patients in Uganda in the SOUTH Study. Clinical Pharmacology & Therapeutics. 2019; 106: 450–457.
29. Mugusi S, Habtewold A, Ngaimisi E, Amogne W, Yimer G, Minzi O, et al. Impact of population and pharmacogenetic variations on efavirenz pharmacokinetics and immunologic outcomes during anti-tuberculosis co-therapy: a parallel prospective cohort study in two sub-Sahara African populations. Frontiers in Pharmacology. 2020; 11: 26.
30. Cerrone M, Wang X, Neary M, Weaver C, Fedele S, Day-Weber I, et al. Pharmacokinetics of efavirenz 400 mg once daily coadministered with isoniazid and rifampicin in human immunodeficiency virus-infected individuals. Clinical Infectious Diseases. 2019; 68: 446–452.
31. Covington N, Luetkemeyer AF, Imperial MZ, Dawson R, Cramer Y, Rosenkranz S, et al. Pharmacogenetics of plasma dolutegravir exposure during 1-month rifapentine/isoniazid treatment of latent tuberculosis. Pharmacogenetics and Genomics. 2025; 35: 140–144.
32. Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research. 2022; 50: D439–D444.
33. Thomas L, Raju AP, Chaithra S, Kulavalli S, Varma M, Sanju SV, et al. Influence of N-acetyltransferase 2 polymorphisms and clinical variables on liver function profile of tuberculosis patients. Expert Review of Clinical Pharmacology. 2024; 17: 263–274.
34. Araujo-Mariz C, Albuquerque MFP, Lopes EP, Ximenes RAA, Lacerda HR, Miranda-Filho DB, et al. Hepatotoxicity during TB treatment in people with HIV/AIDS related to NAT2 polymorphisms in Pernambuco, Northeast Brazil. Annals of Hepatology. 2020; 19: 153–160.
35. Sundell J, Bienvenu E, Janzén D, Birgersson S, Äbelö A, Ashton M. Model-based assessment of variability in isoniazid pharmacokinetics and metabolism in patients co-infected with tuberculosis and HIV: implications for a novel dosing strategy. Clinical Pharmacology & Therapeutics. 2020; 108: 73–80.
36. Kasamatsu A, Miyahara R, Yoneoka D, Toyo-Oka L, Chiyasirinroje B, Imsanguan W, et al. One-year mortality of tuberculosis patients on isoniazid-based treatment and its association with rapid acetylator NAT2 genotypes. International Journal of Infectious Diseases. 2025; 155: 107895.
37. Kengo A, Gausi K, Nabisere R, Musaazi J, Buzibye A, Omali D, et al. Unexpectedly low drug exposures among Ugandan patients with TB and HIV receiving high-dose rifampicin. Antimicrobial Agents and Chemotherapy. 2023; 67: e0043123.
38. Saukkonen JJ, Duarte R, Munsiff SS, Winston CA, Mammen MJ, Abubakar I, et al. Updates on the treatment of drug-susceptible and drug-resistant tuberculosis: an official ATS/CDC/ERS/IDSA clinical practice guideline. American Journal of Respiratory and Critical Care Medicine. 2025; 211: 15–33.
39. Enimil A, Antwi S, Yang H, Dompreh A, Alghamdi WA, Gillani FS, et al. Effect of first-line antituberculosis therapy on nevirapine pharmacokinetics in children younger than three years old. Antimicrobial Agents and Chemotherapy. 2019; 63: e00839-19.
40. Chaivichacharn P, Avihingsanon A, Manosuthi W, Ubolyam S, Tongkobpetch S, Shotelersuk V, et al. Dosage optimization of efavirenz based on a population pharmacokinetic–pharmacogenetic model of HIV-infected patients in Thailand. Clinical Therapeutics. 2020; 42: 1234–1245.
41. Gausi K, Wiesner L, Norman J, Wallis CL, Onyango-Makumbi C, Chipato T, et al. Pharmacokinetics and drug–drug interactions of isoniazid and efavirenz in pregnant women living with HIV in high TB incidence settings: importance of genotyping. Clinical Pharmacology & Therapeutics. 2021; 109: 1034–1044.
42. Dutra CA, Teixeira RLDF, Lopes MQP, Silva VM, Suffys PN, Carvalho RS, et al. Determination of NAT2 genotypes in a cohort of patients with suspected TB in the state of Rio de Janeiro. Pharmaceutics. 2024; 16: 917.
43. Cheng F, Jiang XG, Zheng SL, Wu T, Zhang Q, Ye XC, et al. N-acetyltransferase 2 genetic polymorphisms and anti-tuberculosis drug-induced liver injury: a correlation study. Frontiers in Pharmacology. 2023; 14: 1171353.
44. Sundell J, Bienvenu E, Åbelö A, Ashton M. Effect of efavirenz-based ART on the pharmacokinetics of rifampicin and its primary metabolite in patients coinfected with TB and HIV. Journal of Antimicrobial Chemotherapy. 2021; 76: 2950–2957.