
Download
Review: Hepatology
The Role of the Liver in Iron Homeostasis and What Goes Wrong?
Ernesto Robalino Gonzaga1, 2, Irene Riestra Guiance1, 2, Richard Henriquez1, 2, Gerri Mortimore3, Jan Freeman3*
1University of Central Florida/HCA Healthcare GME, Greater Orlando, FL, USA;
2Department of Internal Medicine, College of Medicine, University of Central Florida, Orlando, FL, USA;
3Hepatology Unit, University Hospitals of Derby and Burton on Trent, Derby, UK
Abstract
Iron is an essential mineral that is vital for growth development, normal cellular function, synthesis of hormones and connective tissue, and most importantly, serves as a component of hemoglobin to carry oxygen to body tissues. The body finely regulates the amount of circulating and stored iron within the body to maintain concentration levels within range for optimal physiologic function. Without iron, the ability for cells to participate in electron transport and energy metabolism decreases. Furthermore, hemoglobin synthesis is altered, which leads to anemia and decreased oxygen delivery to tissue. Problems arise when there is too little or too much iron. This review explores the role of the liver in iron physiology, iron overload and discusses the most common causes of primary and secondary hepatic iron overload.
Key words: liver, iron homeostasis, iron overload
Author for correspondence: Jan Freeman, University of Central Florida/HCA Healthcare GME, Greater Orlando, FL, USA, Email: j.freeman115@btinternet.com
Received: 25 May 2021; Accepted after Revision: 30 June 2021; Published: 18 September 2021
How to cite: Gonzaga ER, et al. The Role of the Liver in Iron Homeostasis and What Goes Wrong? J Ren Hepat Disord. 2021;5(2): 26–33.
Doi: http://dx.doi.org/10.15586/jrenhep.v5i2.110
Copyright: Gonzaga ER, et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0
Introduction
Iron is an essential mineral vital for growth development, normal cellular function, synthesis of hormones, and connective tissue and serves as a component of hemoglobin to carry oxygen to body tissues (1). The body finely regulates the amount of circulating and stored iron to maintain concentration levels within range for optimal physiologic function (2,3). Without iron, the ability of the cells to participate in electron transport and energy metabolism decreases. Furthermore, hemoglobin synthesis is altered, which leads to anemia and reduced oxygen delivery to tissue.
Problems arise when there is too little or too much iron. In excess, iron is toxic, as free-flowing iron can form free radicals via oxidation-reduction reactions (2). Over time, iron excess causes long-term damage to the liver, heart, pancreas, pituitary, joints, and skin. Effects of iron overload (IO) are often insidious and not noticed until widespread tissue damage occurs, leading to irreversible changes such as cirrhosis, diabetes, and arthropathies (3). There are many causes of iron overload including genetic disorders, nonalcoholic liver disease, and responses to acute systemic inflammation or infections.
This review explores the role of the liver in iron physiology, iron overload and discusses the most common causes of primary and secondary hepatic iron overload.
Iron Absorption
The acquisition of iron in a healthy adult is obtained solely through the daily dietary intake (1). In western diets, meats and green leafy vegetables provide an adequate amount of iron to avoid depletion. Upon consumption of an iron-containing substance, iron is converted from ferric iron (Fe3+) to ferrous iron (Fe2+) by intestinal ferric reductase and duodenal cytochrome b(dcytb) reductase (4). An increase in acidity at the enterocyte’s brush border provides a hydrogen electrochemical gradient that drives the ferrous form of iron into the cell. Following uptake, Fe2+ is subsequently transported from the enterocyte’s luminal side into the epithelial cell through either a heme transporter (4), endosome, and/or a divalent metal transport 1 (DMT 1) (2,3,5,6).
If the body requires iron, it is transported to the basolateral membrane by ferroportin (FPN)-1 in association with hephaestin and plasma ceruloplasmin (7). Once iron interacts with hephaestin and gets converted into the ferric state, the state needed for absorption, the iron molecule binds to plasma transferrin. If iron is not required, it becomes sequestered and stored within the cell and expelled along with the enterocyte shedding at the end of its cellular life.
In contrast, heme-iron crosses the enterocyte border without the need for reduction to the ferric state. It is hypothesized that iron is released from heme within the enterocyte and is similarly exported into the bloodstream via FPN-1 (4,7). The body recognizes “need” by the function of ferroportin, which is negatively regulated by the regulatory hormone-hepcidin.
Iron Uptake
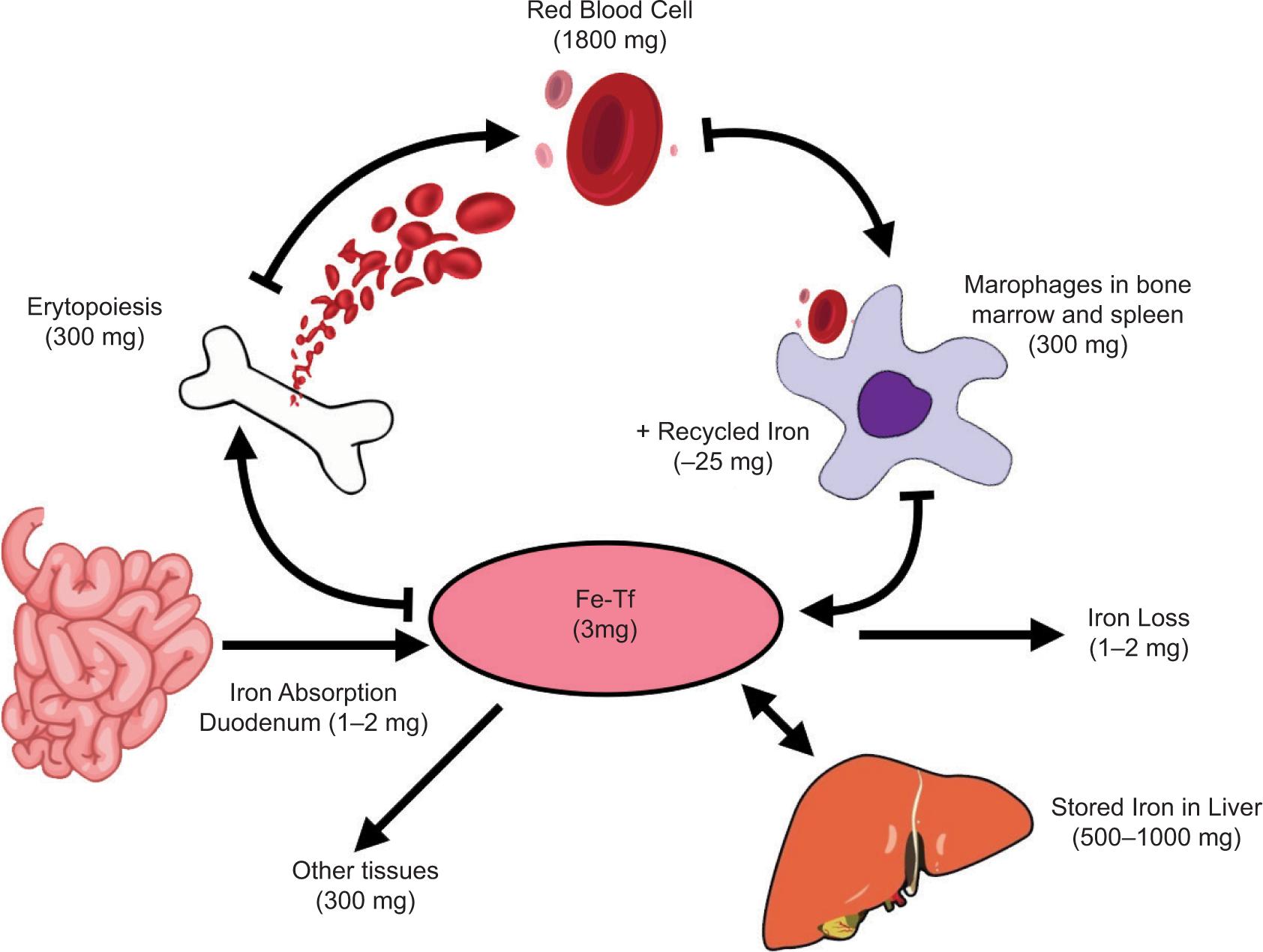
Iron alone is extremely reactive. If iron is not bound by specific serum carriers and/or storage proteins within the body, it can viciously interact with vascular, cellular, and subcellular structures (8). Therefore, after absorption, it is bound to the plasma protein transferrin (TF) for safe transport. TF is a bilobed glycoprotein that consists of two binding sites for iron (2,3,9). Plasma protein affinity allows only five Fe3+ iron molecules to be present at any given time in a healthy adult bloodstream (4). This high affinity prevents free iron from surviving in the bloodstream (4). Iron is then distributed throughout the body to tissues and stored within cells as Ferritin ( Figure 1) (2,3,5,9).
Figure 1: Iron metabolism and distribution.
TF circulates until it interacts with a specific receptor called the transferrin receptor (TfR), which is found on cells with the highest iron demands. These cells include red blood cells (RBCs) and other rapidly dividing cells (4). Even though transferrin receptors are found on many additional cell types around the body, the highest are found in a developing erythroblast. TfR allows the iron molecules to bind and transport iron intracellularly (4). The transferrin-iron complex exists in two forms: monoferric (consists of one iron molecule) or diferric (presence of two iron molecules). In an intelligent physiologic fashion, the TfR has a higher affinity for the diferric form. In contrast, TfR has the lowest affinity for apo TF, which does not carry iron (4).
Once iron binds to TF, the complex travels to the designated tissue to interact with its receptor (10). The TF-TfR complex forms a cluster in the cell membrane on the cell surface and is later internalized as a clathrin-coated vesicle (10). Once in the cytosol, this vesicle will fuse with the endosome to release iron from TF under acidic conditions (10). Acidification is a crucial step for the conformational protein change to release the iron molecule. However, DMT-1 must reduce the iron molecule to its ferrous form to be transported out of the endosome (6,10), which is accomplished by an antigen called six-transmembrane epithelial antigen of the prostate 3 (10). Following this release, the now apo TF, without iron and Tfr complex stays coupled until it returns to the cellular membrane where the pH is more neutral (10). At this stage, the complex will separate, and the apo TF will re-enter the plasma to search for more iron, and the cell can now utilize the released intracellular iron.
Within an erythroblast, any excess iron formed during hemoglobin synthesis gets converted into “ferritin.” This storage mechanism also occurs in other cells, specifically within the liver parenchymal cells, and plays a crucial role in incorporating iron into heme-containing enzymes (4). Iron already incorporated into hemoglobin will enter the human circulation as new RBCs released from the bone marrow. The iron will become part of that red cell mass and cannot be reutilized until the RBC dies (4). At the end of the RBCs life span, the reticuloendothelial (RE) system recognizes senescent cells (4). Here it undergoes phagocytosis, with the hemoglobin forming globin and heme (10).
The resultant iron is then shuttled back to the RE cell’s surface, which TF can recognize, and the cycle restarts anew (10).
Iron Regulation
In a healthy adult, about 30% of TF binding sites are utilized (2,3). Many unoccupied binding sites (~70%) serve as an essential buffer in improper or excess iron absorption through the gastrointestinal tract. If TF binding sites become saturated, as occurs in iron overload disorders, iron is left unbound in its free form, known as non-transferrin-bound iron (NTBI). High NTBI levels are problematic, as the cells and parenchyma that line the body’s organs have no defense against the effect of free iron (2,3,5,9). The body recognizes when TF saturation is becoming high, thus activating a feedback mechanism to signal the body to decrease iron absorption.
On average, the human body maintains approximately 4 g of iron (11). It is estimated that the body absorbs 10% of 10–20 mg of dietary iron (1–2 mg daily) through the gastrointestinal tract. Subsequently, in a cyclical hemostatic fashion, the body also loses old iron at a rate of 1–2 mg/day through sweating, urine, and feces (12). Apart from diet, the body can also accumulate iron through iatrogenic causes, such as excess oral iron supplementations, erythrocyte infusions, or iron transfusions for treatment of certain medical conditions (13,14). The question arises, how is iron metabolism regulated? The answer to this is complicated, and it lies at the biochemical level involving cytoplasmic mRNA. For diferric transferrin to couple with TfR-1, this receptor needs to be synthesized by the induction and sensing of iron deficiency (4). Normally excess iron inhibits TfR-1 synthesis by destabilizing its mRNA through the interaction of iron regulatory protein (IRP)-1.
More importantly, the synthesis of iron receptors and absorption is regulated further by a liver-derived peptide, hepcidin, which functions as an iron regulatory hormone. When bodily iron demands are high, hepcidin levels are low and vice versa (12). When iron absorption is not required, hepcidin is released and binds to FPN-1 to inhibit the further transportation of iron through enterocytes. Hepcidin is upregulated by a protein found on the surface of the liver and produced by the called human homeostatic iron regulator (HFE) gene called HFE protein. They are expressed in response to high circulating iron levels and response to inflammation, infection, endotoxin, and p53 (2,3,9,12). HFE protein is a critical player in iron regulation and the likely culprit of dysfunction in most iron regulatory disorders.
Hepcidin and iron sensing/signaling pathway
Body iron sensing is regulated by a signaling pathway. Hepcidin is a peptide discovered by researchers investigating antimicrobial peptides in the early 2000s (15). Researchers were able to isolate hepcidin from human urine and was named from the basis of its actual synthesis (Hep-) and its antibacterial properties (-cidin) (15,16). It became evident that the peptide played a much larger and essential function in ensuring and maintaining the body’s proper serum iron levels. Although iron overload has been a recognized disorder since the 1900s, the disorder’s mechanism was unknown until hepcidin’s discovery (15). Today, hereditary hemochromatosis is now attributed to hepcidin deficiency.
Hepcidin’s primary mechanism is to act as a negative regulator of ferroportin, the principal iron regulatory hormone (4,17). This protein is synthesized as an 84-amino acid precursor, broken down to its active form, consisting of 25-amino acids (16). In humans, this peptide is derived from the C-terminus end of the prepropeptide, encoded by a 0.4-Kilobase (kb) mRNA generated from three exons of a 2.5 kb gene on chromosome 19 (4). When hepcidin inhibits ferroportin, it prevents the cells from exporting iron back into the blood. Ferroportin is located on the basal membrane of enterocytes and apical membrane of hepatocytes (4,17). Therefore, it will bind to ferroportin and decrease its functional activity through endocytosis from the cell surface and degrade it (4). Hepcidin also functions within the reticuloendothelial cells, the primary storage depot for iron, and inhibits the release of iron recycled from digested RBCs or mobilized from storage pools to cells that need it (4,17).
Hepcidin, ferritin, and TF are acute phase reactants. Iron plays a pivotal role in proliferating, feeding, and fueling the spread of harmful bacteria; ferritin and hepcidin increase to alleviate the amount of readily available serum iron, thus becoming positive acute phase reactants in response to inflammation (4). In contrast, TF is a negative acute phase reactant, lessening in response to inflammation to decrease active iron transportation throughout the blood (4).
Iron concentrations are influenced by multiple stimuli, such as iron body storage, erythropoiesis, hypoxia, and inflammatory states (16). During the elevated iron storage in the body, the levels of hepcidin increase to decrease the amount of iron absorbed (4). Whereas in situations like acute blood loss, hepcidin levels alleviate, and the bioavailable iron will increase. Similarly, in tissue hypoxia, the decreased oxygen will trigger hepcidin production in the liver (4).
Serum ferritin and TF receptor concentrations are altered in response. For example, following surgery, without preceding infection, changes in these acute phase reactants’ concentrations can be observed and are like how the body responds to inflammation because of an infectious process. Iron sensing can be triggered by two signals belonging to transforming growth factor β (TF saturation and bone morphogenic proteins [BMP]) that regulate hepcidin expression. These signals are the extent of TF saturation and bone morphogenic proteins (BMP), a class of ligands of the transforming growth factor β superfamily.
Causes and genetics of iron overload
As discussed, the human body has several regulatory mechanisms to protect itself against the damage caused by free iron. However, in some disorders, these regulatory mechanisms are insufficient or fail, allowing free iron to overload our system (18). When overload occurs, some target organs such as the heart, liver, endocrine cells, gonads, and pancreatic islets are more vulnerable (18). The disorders that cause IO can be separated into mutations that cause an increase in iron absorption via hepcidin dysfunction and mutations that cause ineffective iron export from storage cells via deficiencies in ferroportin or ceruloplasmin (19,20). Commonly, it is a gene mutation and a combination of environmental factors, particularly alcohol intake of more than 60 g/day, coupled with an underlying gene mutation that ultimately contributes to the development of clinically relevant IO (21).
The most common IO disorders are those that encompass hereditary hemochromatosis. There are four types of hemochromatosis. Three are recessive mutations affecting the activation of hepcidin via HFE, HJV, HAMP, and TRF2. Type 4 hemochromatosis/ ferroportin disease is an autosomal dominant mutation in the hepcidin receptor FPN (22). The HFE protein is regulated through TfR-1 binding and endocytosis of diferric transferrin control synthesis (4). TfR-1 binds to HFE using a similar binding site as holotransferrin. When holotransferrin is elevated, it will displace the HFE from the TfR1 and allows HFE to signal the liver to increase hepcidin transcription (4).
Moreover, the HFE gene, protein, and hepcidin signaling are directed by the needed erythropoiesis requirement. Overall, the liver regulates hepcidin transcription reduction, allowing iron to be absorbed according to the body’s demands (4). Therefore, mutation of one of the genes involved with hepcidin or hepcidin results in extreme iron overload from uncontrolled iron absorption from the intestine.
The commonly occurring hemochromatosis is type 1 HFE-related hemochromatosis. Typically caused by a mutation of cysteine-to-tyrosine substitution at amino acid 282 (C282Y), which carries a low clinical penetrance with variable expression (16). This mutation causes an inactivation in hepcidin, leading to dysregulation of iron absorption by enterocytes. If clinically meaningful, symptoms appear between the ages of 40 and 60 years. A mutation in the SLCS401A1 gene causing ineffective iron export and a complete loss of ferroportin’s iron export function is responsible for Type 4 hemochromatosis. This loss of function causes iron to accumulate in macrophages; thus, upregulating ferritin and iron release into plasma. Because the macrophages can handle the excess iron, it does not cause any clinically significant disease. The most lethal subtypes are Type 2A and 2B, caused by HJV and HAMP mutations, as they are known to be primarily responsible for lower hepcidin levels (23).
Other IO syndromes include atransferrinemia, DMT-1 deficiency, and aceruloplasminemia. Atransferrinemia is caused by a pathogenic mutation leading to low serum TF causing poor iron delivery to mature erythroblasts (6,16). DMT1 is a transmembrane protein in the brush-border membrane of duodenal enterocytes and plays a role in the use of iron by erythroid precursors (6,16,24). A mutation in the coding of the protein is infrequent. Acerulopasminemia is an extremely rare mutation within the multicopper ferroxidase family that is responsible for facilitating the efflux of iron and its release to TF with variable expression. This mutation is exceptionally toxic to neurons and is the only IO disorder with neurological manifestations (6,19,24).
The secondary causes of IO include ineffective erythropoietic disorders such as thalassemia syndromes, sideroblastic anemia, myelodysplastic syndrome, parental iron overload, testosterone use, and end-stage liver disease (25). The causes of IO are usually because of the need for multiple iron transfusions along with increased absorption of iron by the intestine to provide more iron to produce erythrocytes (25).
Identifying symptoms and diagnosis
Regardless of the IO disorder etiology, severe iron overload leads to clinically significant IO disorder, characterized by deposition of hemosiderin in the liver and other organs. Clinical manifestations are linked to the iron accumulation in the tissues and the organ’s ability to cope with oxidative management. Typically, organ damage is more commonly evident in males versus females, presumably because of menstruation. Type 1 hemochromatosis carries the highest risk for cirrhosis and a two-fold risk for hepatocellular carcinoma, and type 1A and type 2B are associated with death because of cardiac dysfunction (26,27).
Hepatic injury is mediated by an inflammatory response by Kupffer cells that lead stellate and mesenchymal cells to produce excess collagen and cause fibrosis. Initially, iron deposition occurs in the cytoplasm of periportal hepatocytes. However, as iron load increases, the rest of the lobule along with the bile duct epithelium and Kupffer cells are affected. About 13 to 15 mg of iron/g of liver tissue is necessary for the development of cirrhosis. Normal individuals have less than 1000 ug/g dry weight of iron within liver tissue. Those with over 22,000 iron ug/g dry weight of iron almost certainly have cirrhosis (28).
Diagnosis is usually incidental during routine labs as IO presents with nonspecific symptomatology that develops slowly. Although opinions vary, the guidelines from 2005–2018 suggest that elevated ferritin of >300 ug/L in males and postmenopausal females and >200 ug/L in premenopausal females with and increased fasting TF saturation >45% with or without symptoms indicate further investigation (11,12,26,27,29). Ferritin overestimates the amount of serum iron and can be influenced by alcohol use, inflammation, infection, metabolic syndrome, and other inflammatory states; thus, it is not a reliable diagnostic index for IO disorders. Hemoglobin, mean corpuscular volume, TF saturation are other biochemical indices that further elucidate the etiology of IO. Increased TF saturation level indicates unusual iron hemostasis because of increased intestinal iron absorption along with iron release from macrophages and other storage cells.
Hyperferritinemia with normal to low TF saturation triggers the identification of other pathologies such as cell necrosis, metabolic syndrome, alcohol use disorder, or mutations causing elevated ferritin levels. Microcytic anemia indicates that the IO disorder responsible is likely DMT1 deficiency, aceruloplasminemia, or atransferrinemia. Overall, liver biopsy is the gold standard in diagnosis and determine the amount of iron overload and degree of liver damage. However, quantitative magnetic resonance is a noninvasive modality to estimate the iron load in organs and is widely used (30). Genetic testing and screening are necessary to confirm the diagnosis and identify at-risk relatives (19).
Individuals with a known family history of hereditary hemochromatosis (HH) should be screened for HFE gene mutations. Individuals who self-reported family history of HH had a sensitivity and specificity of 81% and 97%, respectively, supporting family history as an accurate screening tool (31).
In those patients who are not aware of their mutation status but have clinical manifestations, diagnosis can be confirmed by combining laboratory and diagnostic tests. Overall, guidelines do not recommend genetic testing amongst the general population. Testing for HFE mutation is not recommended because of its low penetrance, which only shows a small percentage of homozygous iron overload in individuals progressing to IO. Patients with unexplained arthritis, arthralgia, or Type 2 diabetes are not recommended to get tested for HH. However, testing should be considered in unexplained chronic liver disease with high serum levels of TF saturation (12,27,32,33).
No further genetic testing is recommended if tested individuals do not carry the C282Y or H63D alleles. Patients with H63D or S65C mutations but lacking the C282Y mutation should be reassured that they are not at higher risk for IO. A biopsy is only recommended for confirmation if needed, to stage liver fibrosis or in the setting of serum ferritin above 1000 mg/L, elevated AST, hepatomegaly, or above 40 years of age. Otherwise, the American College of Gastroenterology (ACG) recommends contrast-enhanced magnetic resonance imaging (MRI) to measure liver iron concentration noninvasively (27).
Treatment
Guidelines agree that phlebotomy is the recommended first-line treatment in HH (26,27,33). Treatment should begin in C282Y homozygotes with elevated serum ferritin of >300 mg/mL in men and >200 mg/mL in women, combined with a serum TF saturation of >45%. Phlebotomy occurs weekly, removing an estimated ~500mL of blood to reduce serum ferritin level to a goal level of 50 to 100 ng/mL. The frequency of phlebotomy occurs on a maintenance schedule of around 3–4 times a year after the goal level achievement (34).
Erythrocytapheresis can be used as an alternative to phlebotomy. This method targets and removes only RBCs, particularly in individuals suffering from hypoproteinemia or thrombocytopenia. It can remove up to 800mL of RBCs in volume during one session and requires less frequent removal every 2–3 weeks. However, the procedure is much more expensive than phlebotomy and is not recommended in routine cases (27).
Chelation therapy is only suggested for patients who are intolerant or refractory to phlebotomies (27). While effective, chelating agents can have adverse effects. The three most common chelating agents: deferoxamine, deferiprone, and deferasirox, have the potential for renal and hepatic toxicity, which requires frequent laboratory monitoring, especially of creatinine, as there is an increased risk for acute renal failure (27). Additionally, these oral agents can also cause gastrointestinal upset, bleeding, pain, and rash, making compliance more difficult. A risk-benefit analysis should be considered when choosing chelation and the specific agent for each patient (35–39).
Proton pump inhibitors (PPIs) can reduce iron absorption by decreasing gastric acid secretion. Several studies have shown a reduction in the number of phlebotomies needed to achieve goal TF levels (40–42). However, contrary to EASL and ASLD guidelines, ACG 2019 recommends against PPI as a primary treatment of HH.
A diet low in iron, alcohol, meat, and vitamin C is recommended with refraining from shellfish or undercooked pork because of an increased risk for infection (22).
Liver transplant should be considered in all patients with HH and decompensated cirrhosis or hepatocellular carcinoma.
Nonalcoholic fatty liver disease (NAFLD) related to iron overload
NAFLD is defined as the presence of hepatic steatosis with no secondary causes of hepatic fat accumulation (i.e., alcohol consumption, hepatitis). NAFLD afflicts patients worldwide but is the most common amongst individuals living in the Western industrialized countries. Based on the severity of hepatic inflammation, NAFLD is subdivided into two categories: the milder NAFLD and the more severe nonalcoholic steatohepatitis (NASH). In NAFLD, there is an accumulation of triglycerides within the hepatocytes in the absence of significant alcohol consumption (43). In contrast, in NASH, fatty build-up causes definitive lobar inflammation and cell death, leading to fibrosis and cirrhosis.
About 33% of patients with NAFLD exhibit evidence of disrupted iron homeostasis (44). These patients often have elevated serum ferritin levels with either normal or mild elevation in TF saturation levels, and additionally, upon liver biopsy, they often have a hepatic iron disposition. The term “dysmetabolic iron overload syndrome (DIOS)” has is used to describe these findings (45).
Iron accumulation in NAFLD is secondary to the inhibition of iron use from hepatocytes (46). Impaired export leads to inflammation and metabolic derangements. That impacts iron regulators (hepcidin and ferroportin), TF receptors, ferritin, and copper.
An increase in the ferritin levels is a crucial key feature of iron dysregulation in patients with NAFLD. There appears to be a correlation between hyperferritinemia levels and the degree of histologic liver injury (46). In a large NASH study, serum ferritin concentration of more than 1.5 times, the upper limit of normal was associated with advanced liver fibrosis and NAFLD activity score (47).
Iron deposition in NALFD is found within the Kupffer cells and hepatocytes. The Kupffer cells are involved in initiating the inflammatory cascade when it responds to the uptake of oxidized lipoproteins and iron accumulation (48). In NAFLD, hepcidin expression was directly correlated with iron liver, indicating that these patients have an intact physiologic response to hepcidin biosynthesis, unlike hemochromatosis (48).
Clinically, the excess iron caused by NAFLD causes similar iron overload to hemochromatosis. Excess unbound iron can catalyze the formation of toxic hydroxyl radicals and can result in lasting cellular damage. Therefore, DIOS can lead to long-term effects of hepatic damage, cardiovascular disease, and predisposing metabolic disorders, such as diabetes. Iron overload may also contribute to liver injury in NAFLD by adding stress to the endoplasmic reticulum within hepatocytes (46). In NAFLD, the excess iron generates an unfolded protein response and stress upon the endoplasmic reticulum within the hepatocytes. Similarly, iron overload upregulates cholesterol pathways, further potentiating NAFLD. Hence efforts to reverse or lessen the iron burden may improve or slow the progression of NAFLD (46).
Treatment for iron overload in NAFLD mainly focuses on reducing hepatic steatosis. That improves liver dysfunction and prevent further fibrosis. Recommendations for NAFLD treatment start with lifestyle changes, including weight loss, refraining from alcohol consumption, and exercise. A weight reduction of at least 5% of an individual’s body weight can improve fatty deposition. In a meta-analysis study, ≥ 5% body weight loss improved fatty liver disease, and loss of ≥ 7% of body weight improved NALFD activity score (NAS) (49). For patients with diabetes and the presence of NASH, treatment should also target lowering glycated hemoglobin.
Hyperferritinemia
Serum ferritin is a well-known positive acute-phase reactant, and its elevation is seen in any stress state. In these states, translation and transcription of ferritin are upregulated, and hepatocytes, Kupffer cells, macrophages, and proximal tubular renal cells are shown to secrete ferritin. Hyperferritinemia is ubiquitous, and it is a marker of multiorgan dysfunction associated with high mortality regardless of its etiology (50). Any disease state with underlying inflammation such as juvenile idiopathic arthritis and even diabetes can lead to hyperferritinemia.
Despite the prevalence of hyperferritinemia, it may or may not be accompanied by increased serum iron. Several hereditary disorders have elevated serum ferritin but no iron overload. That include familial hemophagocytic lymphohistiocytosis, hereditary hyperferritinemia cataract syndrome, and Gaucher’s disease (51–54).
Conclusion
Iron is an essential mineral vital to the body for normal cellular functioning and growth. Maintaining proper iron concentrations is crucial for optimal physiologic function (2,3). Excess build-up of iron is dangerous and can lead to irreversible organ dysfunction. Numerous regulatory entities that take part in iron homeostasis levels, but the protein hepcidin seems to be one of the largest culprits for abnormal regulation and overload. Iron overload disorders, such as HH, are often insidious and difficult to diagnose. However, successful treatment options exist and can prevent damage, allowing an individual to evade iron’s troublesome effects.
Conflicts of Interest
No potential conflicts of interest were reported by the authors.
REFERENCES
1. Office of dietary supplements - Iron [Internet]. [Updated
2. Chipman J, Laubenbacher R, Torti S. A systems biology approach to iron metabolism. In: Corey, S, Kimmel M, Leonard J, editors. A systems biology approach to blood. Advances in experimental medicine and biology. Vol 844. New York, NY: Springer; 2014. p. 201–25. 10.1007/978-1-4939-2095-2
3. Ded S, Babbitt J. Overview of iron metabolism in health and disease. Hemodial Int. 2017; 21:S6–20. 10.1111/hdi.12542
4. Janson LW, Tischler ME. The big picture. Medical biochemistry. New York: McGraw-Hill; 2012.
5. Milic S, Mikolasevic I, Orlic L, et al. The role of iron and iron overload in chronic liver disease. Med Sci Monit Basic Res. 2016;22:2144–51. 10.12659/MSM.896494
6. Lolascon A, De FL. Mutations in the gene encoding DMT 1: Clinical presentation and treatment. Semin Hematol. 2009;46(4):358–70. 10.1053/j.seminhematol.2009.06.005
7. Pietrangelo A. Ferroportin disease: Pathogenesis, diagnosis and treatment. Haemotologica. 2017 Dec;102(12):1972. 10.3324/haematol.2017.170720
8. Boyer TD, Manns MP, Sanyal AJ, Zakim D. Zakim and Boyer’s hepatology: A textbook of liver disease. Philadelphia, PA: Saunders/Elsevier; 2012.
9. Bloomer S, Brown K. Iron-induced liver injury: A critical reappraisal. Int J Mol Sci. 2019;20(9):21–32. 10.3390/ijms20092132
10. Ganz T Iron metabolism. In: Kaushansky K, Lichtman MA, Prchal JT, et al., editors. Williams hematology. 9th ed. New York: McGraw-Hill; 2015.
11. Fitzsimons E, Cullis J, Thomas D, et al. Diagnosis and therapy of genetic haemochromatosis (review and 2017 update). Br Haematol. 2018;181(3):293–303. 10.1111/bjh.15164
12. Katsarou M, Papasavva M, Latsi R, Drakoulis, N. Hemochromatosis: Hereditary hemochromatosis and HFE gene. Vitam Horm. 2019;110:201–22. 10.1016/bs.vh.2019.01.010
13. De Sanctis V. A young adult with unintentional acute parenteral iron intoxication treated with oral chelation: The use of liver ferriscan. Mediterr J Hematol Infect Dis. 2016;9(1):e2017008. 10.4084/mjhid.2017.008
14. De Sanctis V. Liver iron content (LIC) in adults with non-transfusion dependent sickle cell disease (NT-SCD). Correlation with serum ferritin and liver enzymes concentrations. Mediterr J Hematol Infect Dis. 2017;9(1):2017037. 10.4084/mjhid.2017.037
15. Rossi E. Hepcidin-the iron regulatory hormone. Clin Biochem Rev. 2005;26(3):47–9. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1240030/
16. Kemna EH, Tjalsma H, Willems HL, et al. Hepcidin: From discovery to differential diagnosis. Haematologica. 2008 Jan;93(1):90–7. 10.3324/haematol.11705
17. Billesbolle CB, Azumaya CM, Kretsch RC et al. Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature. 2020 Oct;586(7831):807–11. 10.1038/s41586-020-2668-z
18. Bunn H, Heeney MM. Iron homeostasis: Deficiency and overload. In: Aster JC, Bunn H, editors. Pathophysiology of blood disorders. 2nd ed. New York: McGraw-Hill; 2016.
19. Piperno A, Pelucchi S, Mariani R. Inherited iron overload disorders. Transl Gastroenterol Hepatol. 2020;5:25. 10.21037/tgh.2019.11.15
20. Armitage AE, Stacey AR, Giannoulatou E, et al. Distinct patterns of hepcidin and iron regulation during HIV-1, HBV and HCV infections. Proc Natl Acad Sci USA. 2014 Aug 19;111(33):12187–92. 10.1073/pnas.1402351111
21. Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet. 2016;388(10045):706–16. 10.1016/S0140-6736(15)01315-X
22. Adams P, Altes A, Brissot P, et al. Therapeutic recommendations in HFE hemochromatosis for p.Cys282Tyr (C282Y/C282Y) homozygous genotype. Hepatol Int. 2018;12(2):83–6. 10.1007/s12072-018-9855-0
23. Ganz T. Iron deficiency and overload. In: Kaushansky K, Lichtman MA, Prchal JT, et al., editors. Williams hematology. 10th ed. New York: McGraw-Hill; 2021.
24. Mims MP, Guan Y, Pospisilova D, et al. Identification of a human mutation of DMT1 in a patient with microcytic anemia and iron overload. Blood. 2005;105(3):1337–42. 10.1182/blood-2004-07-2966
25. Kennelly PJ, Murray RK, Jacob M, Varghese J. Plasma proteins & Immunoglobulins. In: Rodwell VW, Bender DA, Botham KM, et al. Harpers illustrated biochemistry. 31 ed. New York: Mcgraw-Hill, 2018.
26. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010;53(1):3–22. 10.1016/j.jhep.2010.03.001
27. Kowdley K, Brown K, Ahn J, Sundaram V. ACG Clinical guideline. Am J Gastroenterol Suppl. 2019;114(8):1202–18. 10.14309/ajg.0000000000000315
28. Kumar V, Abbas AK, Aster JC. Robbins and Cotran pathologic basis of disease. Philadelphia, PA: Elsevier/Saunders; 2015.
29. Qaseem A, Aronson M, Fitterman N, et al. Screening for hereditary hemochromatosis: A clinical practice guideline from the American College of Physicians. Ann Intern Med. 2005;143(7):517. 10.7326/0003-4819-143-7-200510040-00010
30. Golfers S, Lewis S, Weisberg IS. Hemochromatosis: Pathophysiology, evaluation, and management of hepatic iron overload with a focus on MRI. Expert Rev Gastroenterol Hepatol. 2018;12(8):767–78. 10.1080/17474124.2018.1496016
31. Acton R, Barton J, Passmore L, et al. Accuracy of family history of hemochromatosis or iron overload: The hemochromatosis and iron overload screening study. Clin Gastroenterol Hepatol. 2008;6(8):934–8. 10.1016/j.cgh.2008.04.003
32. Beutler E, Felitti VJ, Koziol JA, et al. Penetrance of 845G → A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet. 2002;359:211–8. 10.1016/S0140-6736(02)07447-0
33. Bacon B, Adams P, Kowdley K, et al. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the study of liver diseases. Hepatology. 2011;54(1):328–43. 10.1002/hep.24330
34. Brissot P. Optimizing the diagnosis and the treatment of iron overload diseases. Expert Rev Gastroenterol Hepatol. 2016;10(3):359–70. 10.1586/17474124.2016.1119043
35. Hoffbrand AV, Taher A, Cappellini MD. How I treat transfusional iron overload. Blood 2012;120:3657. 10.1182/blood-2012-05-370098
36. Lee JW, Yoon SS, Shen ZX, et al. Iron chelation therapy with deferasirox in patients with aplastic anemia: A subgroup analysis of 116 patients from the EPIC trial. Blood 2010;116:2448. 10.1182/blood-2010-01-261289
37. Boxed warning about deferasirox [Internet]. [Cited
38. Tanner MA, Galanello R, Dessi C, et al. A randomized, placebo-controlled, double-blind trial of the effect of combined therapy with deferoxamine and deferiprone on myocardial iron in thalassemia major using cardiovascular magnetic resonance. Circulation. 2007;115:1876. 10.1161/CIRCULATIONAHA.106.648790
39. Kowdley KV, Kaplan MM. Iron-chelation therapy with oral deferiprone--toxicity or lack of efficacy? N Engl J Med. 1998;339:468. 10.1056/NEJM199808133390709
40. Van Aerts RM, van Deursen CT, Koek GH. Proton pump inhibitors reduce the frequency of phlebotomy in patients with hereditary hemochromatosis. Clin Gastroenterol Hepatol. 2016;14(1):147–52. 10.1016/j.cgh.2015.06.043
41. Vanclooster A, van Deursen C, Jaspers R, et al. Proton pump inhibitors decrease phlebotomy need in HFE hemochromatosis: Double-blind randomized placebo-controlled trial. Gastroenterology 2017;153(3):678–80.e2. 10.1053/j.gastro.2017.06.006
42. Hutchinson C, Geissler CA, Powell JJ, et al. Proton pump inhibitors suppress absorption of dietary non-haem iron in hereditary haemochromatosis. Gut. 2007;56(9):1291–5. 10.1136/gut.2006.108613
43. Cohen DE, Anania FA. Nonalcoholic fatty liver disease. In: Greenberger NJ, Blumberg RS, Burakoff R, editors. Current diagnosis & treatment: Gastroenterology, hepatology, & endoscopy. 3rd ed. New York: McGraw-Hill; 2016.
44. Williams CD, Stengel J, Asike MI, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology. 2011;140:124. 10.1053/j.gastro.2010.09.038
45. Datz C, Müller E, Aigner E. Iron overload and nonalcoholic fatty liver disease. Minerva Endocrinol. 2017 Jun;42(2):173–83. 10.23736/S0391-1977.16.02565-7
46. Britton LJ, Subramaniam VN, Crawford DH. Iron and nonalcoholic fatty liver disease. World J Gastroenterol. 2016;22(36): 8112–22. 10.3748/wjg.v22.i36.8112
47. Sumida Y, Yoneda M, Hyogo H, et al. A simple clinical scoring system using ferritin, fasting insulin, and type IV collagen 7S for predicting steatohepatitis in nonalcoholic fatty liver disease. J Gastroenterol. 2011;46:257–68. 10.1007/s00535-010-0305-6
48. Aigner E. Dysregulation of iron and copper homeostasis in nonalcoholic fatty liver. World J Hepatol. 2014;7(2):177. 10.4254/wjh.v7.i2.177
49. Musso G, Cassander M, Rosina F, Gambino R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in nonalcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of randomised trials. Diabetologia. 2012;55(4):885–904. 10.1007/s00125-011-2446-4
50. Bennett TD, Hayward KN, Farris RW, et al. Very high serum ferritin levels are associated with increased mortality and critical care in pediatric patients. Pediatr Crit Care Med. 2011;12(6):e233–6. 10.1097/PCC.0b013e31820abca8
51. Ravasi G, Pelucchi S, Mariani R, et al. Unexplained isolated hyperferritinemia without iron overload. Am J Hematol. 2017;92(4):338–43. 10.1002/ajh.24641
52. Kernan KF, Carcillo JA. Hyperferritinemia and inflammation. Int Immunol. 2017;29(9):401–9. 10.1093/intimm/dxx031
53. Al-Samkari H, Berliner N. Hemophagocytic lymphohistiocytosis. Annu Rev Pathol. 2018;13(1):27–49. 10.1146/annurev-pathol-020117-043625
54. Cadenas B, Fita-Torró J, Bermúdez-Cortés M, et al. L-Ferritin: one gene, five diseases; from hereditary hyperferritinemia to hypoferritinemia—Report of new cases. Pharmaceuticals. 2019;12(1):17. 10.3390/ph12010017