
Download
Case Reports: Nephrology
A Neglected Case of Wilson’s Disease Presenting as Symptomatic Urolithiasis and Proteinuria: A Case Report and Review of the Literature
Elham Zare1*, Zahra Mahbubi2, Maryam Panahi3
1Department of Internal Medicine, North Khorasan University of Medical Sciences, Bojnurd, Iran;
2Department of Radiology, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran;
3School of Medicine,Tehran University of Medical Sciences, Tehran, Iran
Abstract
We report a short-statured, 39-year-old male presenting with recurrent kidney stones, history of refractory rickets, and bone deformity. He had been consuming multiple doses of calcium supplements and multiple courses of vitamin D over past 30 years prior to reporting in our clinic without any significant laboratory or clinical improvement. The patient was diagnosed as having Fanconi’s syndrome attributable to Wilson’s disease. This patient highlighted that in case of resistant rickets, a high index of uncertainty must be invoked for Wilson’s disease. Appropriate timely recognition of this entity results in prompt ministrations and prevention of disability. We also presented and discussed reviews on Wilson’s disease from literature.
Key words: bone disease, renal tubular acidosis, Wilson’s disease
Received: 20 August 2021; Accepted after Revision: 3 September 2021; Published: 7 October 2021
Author for correspondence: Department of Internal Medicine, North Khorasan University of Medical Sciences, Bojnurd, Iran. Email: elhamzr56@gmail.com
How to cite: Zare E, et al. A Neglected Case of Wilson’s Disease Presenting as Symptomatic Urolithiasis and Proteinuria: A Case Report and Review of the Literature. J Ren Hepat Disord. 2021;5(2): 38–43.
Doi: http://dx.doi.org/10.15586/jrenhep.v5i2.123
Copyright: Zare E, et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0
Introduction
Vitamin D deficiency because of inadequate serum levels is the most common cause of rickets that becomes apparent in the early years (1).
A comprehensive inspection of other etiologies of rickets is suggested in patients who are intractable to the replacement of vitamin D, including hypophosphatemic rickets, renal tubular acidosis (RTA), renal osteodystrophy, and vitamin D-resistant rickets (1–5).
Although infrequent, Fanconi’s syndrome is a significant cause of hypophosphatemic rickets. It is described as proximal RTA when presented with other proximal tubular defects such as aminoaciduria, phosphaturia, glucosuria, and uricosuria (6).
Fanconi’s syndrome occurs either as a primary or secondary type and causes derangement in the functioning of renal tubular cells. Some toxic metabolic substances also cause this syndrome such as in Wilson’s disease.
Wilson’s disease is an inherited disease. Generally, it presents with hepatic and neuropsychiatric expressions because of excessive deposition of copper in organs due to defect of copper transport by hepatic lysosomes (7). Extrahepatic manifestations, including renal and osteoarticular disorders, although described infrequently, present as features of Wilson’s disease (8–10).
We present a 38-year-old male with rickets refractory to replacement of vitamin D because of Fanconi’s syndrome secondary to Wilson’s disease. A written informed consent was obtained from the patient, who was informed that the data could be submitted for publication.
Case Report
A 39-year-old male, third born from a second consanguineous marriage, was referred to our clinic for evaluation of proteinuria. He had a history of recurrent kidney stone formation, with long-term generalized weakness, pain in bones, progressive struggling in walking, and emergence of bone deformity in the extremities.
According to the patient, the stones passed with conservative measures but were neither retrieved nor were any biochemical studies conducted.
His symptoms appeared at about 10 years of age with pain in bones. He was unable to walk properly since adulthood. He had bowing of the legs, which started in childhood and had gradually progressed to the present status. He was not able to walk without support for the past 15 years.
He was unable to attend and finish schooling after 8th grade. There was no history of involuntary movement, seizures, or polyuria.
He had been treated with multiple courses of calcium supplements and vitamin D for past 30 years before reporting to our clinic but without any notable clinical or testing improvement.
One of his brothers had died at the age of 40 after rapid evolution of jaundice and severe ascites, but no conclusive diagnosis was made at that time.
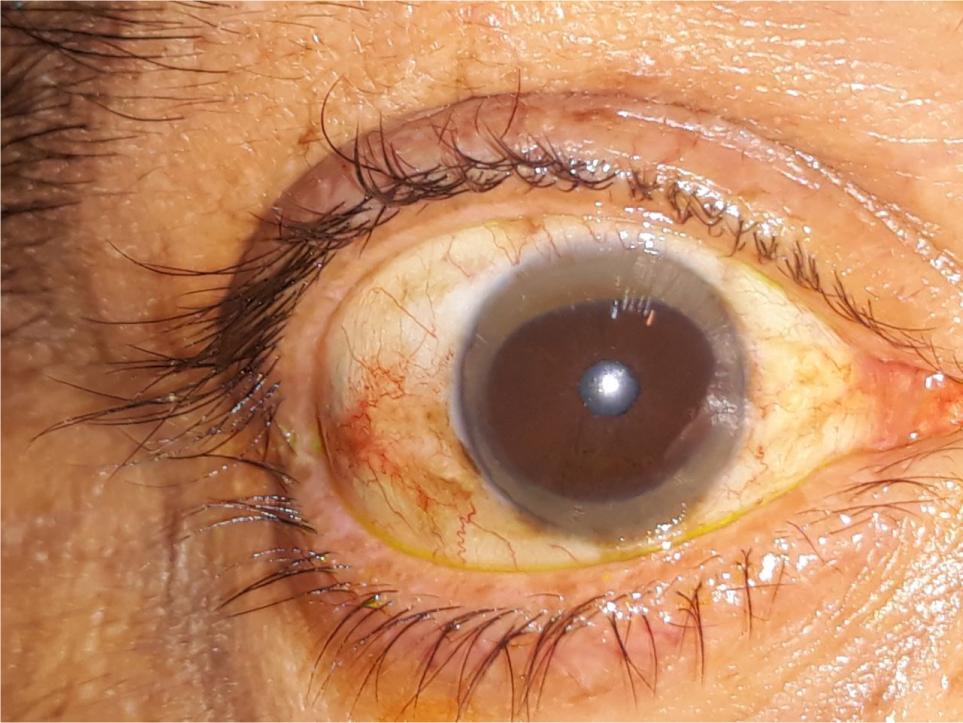
General physical examination of our patient presented non-icteric, well-oriented male with severe short stature, height being 141 cm and weight as 45 kg. Vital signs were stable. Ophthalmic examination depicted presence of Kayser–Fleischer (K-F) rings (Figure 1), which was later confirmed by ophthalmologist on slit lamp examination.
Figure 1: Ophthalmic examination shows presence of Kayser–Fleischer (K-F) rings.
The abdomen was protuberant without any organomegaly. Secondary sexual characters were well developed.
All muscle groups were atrophied. Rachitic changes were noted, which included lordosis, genu valgum, prominent costo-chondral junction, and several bone deformities. The bones were tender on palpation.
Neurological examination had no sign of cerebellar dysfunction, including extra pyramidal involvement and ataxia. The motor system evidenced normal reflexes and tones, muscle force was 3/5 in the lower limbs and 4/5 in the upper limbs.
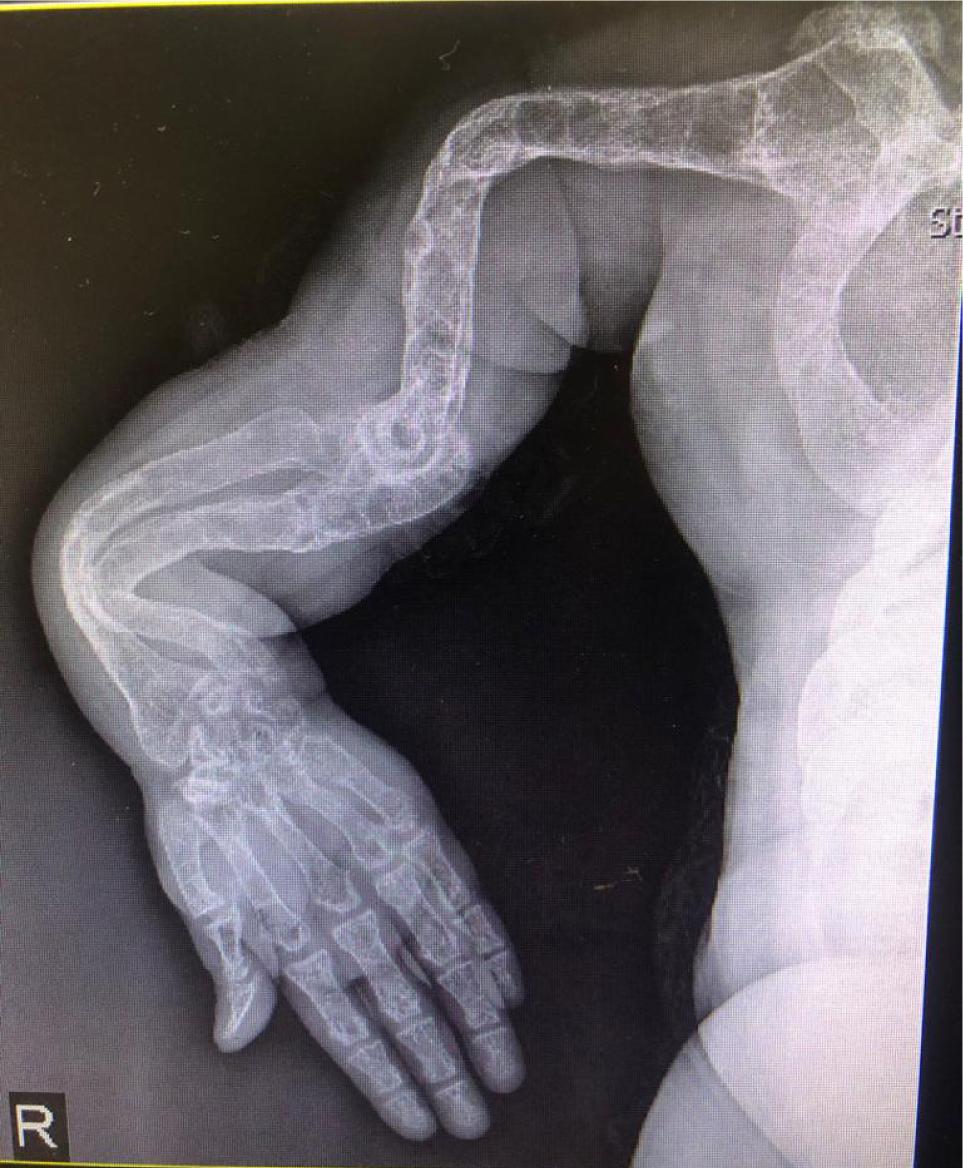
Clinical examination established diagnosis of rickets. The patient depicted severe bowing of the upper and lower limbs and marked generalized osteopenia (Figure 2). Bony fusion and significant osteoarthritis was observed in the larger joints, with apparent presence in his knees. Angulation of the carpal bones and squaring in the head of the metacarpal bones was demonstrated. Severe deformity of the rib cage, inward bowing of the ribs, and marked kyphoscoliosis were also analyzed.
Figure 2: X-ray reveals severe bowing of the upper and lower limbs and marked generalized osteopenia.
Laboratory investigations
Laboratory analyses revealed the following results: level of 25-hydroxyvitamin D [25(OH)D] was adequate (52 ng/dL), with normal corrected calcium (10.24 mg/dL), phosphate (2.6 mg/dL), albumin (3.8 g/dL), blood Urea (17.7 mg/dL), creatinine (0.6 mg/dL), creatinine phosphokinase (59 IU/L), lactate dehydrogenase (426 IU/L), sodium (143 mEq/L), potassium (3.4 mEq/L; normal range: 3.6–5.0 mEq/L).
Further analyses depicted the following values: white blood corpuscle (WBC) = 7100/mm3, hemoglobin = 16 g/dL, mean corpuscular volume (MCV) = 87.8 Fl, mean corpuscular hemoglobin (MCH) = 30.5, mean corpuscular hemoglobin concentration (MCHC) = 34.7, platelet count = 138,000/mm3, urine analysis (specific gravity = 1005, glycosuria = 2+ [corresponding blood sugar was normal], proteinuria = 1+, triple phosphate crystals present, and pH = 8), and C-reactive protein (CRP) = negative. The erythrocyte sedimentation rate (ESR) was 3 mm in the first minute, prothrombin time = 14.7 s, partial thromboplastin time = 35 s, SGOT = 66 U/L, SGPT = 54 U/L, bilirubin total = 2.04 mg/dL, direct bilirubin = 0.63 mg/dL, serum alkaline phosphatase levels were elevated at 312 IU/L, and lactate dehydrogenase (LDH) = 496 U/L.
Further laboratory investigations revealed that viral serologies for human immunodeficiency virus (HIV), hepatitis B antigen, and anti-hepatitis C virus Ab were negative. Upper gastrointestinal endoscopy was also within normal limits. The 24-h urinalysis established the following parameters: urine volume = 2000 cc; creatinine = 380 mgl(for males 97 to 137 ml/min); protein = 860 mg/day(less than 80 mg per 24 hrs); calcium = 95 mg/day (normal range: 100–300 mg/day); uric acid = 228 mg/day (normal range: 250–750 mg/day); phosphate = 0.729 mg/day (normal range: 400–1300 mg/day); and magnesium = 45 mg/day (normal range: 71–121 mg/day.
Further tests presented antismooth muscle antibodies (ASMA) = negative, antinuclear antibodies (ANA) = 0.4 U/mL, anti-liver–kidney microsomal antibody (LKM Ab) = 0.1 U/mL, thyroid function tests were normal, transferrin saturation = 43.7%, ferritin = 139 ng/mL, parathormone = 53 pg/mL (normal range: 13–54), arterial blood gas depicted a pH of 7.25, partial pressure of carbon dioxide (pCO2) = 22 mm Hg, serum chloride = 117 mEq/L (normal range up to 110), serum bicarbonate = 13.8 mEq/L, urine chloride = 135 mEq/L (98–107 mEq/L), urine potassium = 28.8 MEq/24 h, urine anion gap = +31.
Capillary serum and urine electrophoresis analysis were normal. Serum copper (40 µg/dL), ceruloplasmin (13 mg/dL) (normal range: 18–35 mg/dL), and 24-h urine copper was 220 mcg (normal value is up to 70 mcg/24 h). Glycosuria (1.83 g/L) was found on urinalysis with normal blood glucose.
Laboratory test data were compatible with normal anion gap RTA. According to the laboratory data, Fanconi’s syndrome due to Wilson’s disease was suspected clinically.
An abdominal ultrasound examination of the patient revealed multiple cortical cysts along with multiple small size stones in his right kidney. The liver parenchyma appeared to be having a heterogeneous and coarse architecture; the outline appeared markedly nodular, suggestive of chronic parenchymal liver disease, that is, liver cirrhosis. The spleen and portal vein were normal. Upper gastrointestinal endoscopy was also normal. However, the ammonium chloride challenge test was positive, indicating proximal tubular dysfunction, and the diagnosis of Wilson’s disease was confirmed after ophthalmological consultation.
The patient was placed on the replacement therapy of calcium, phosphate, and vitamin D along with replacement of alkali. He was also started with zinc acetate, 50 mg three times a day, and D-penicillamine, 250 mg/day, which was gradually increased to 750 mg/day given in divided doses, along with 20 mg/day of oral pyridoxine.
Discussion and Review of Literature
In 1912, Wilson (11) reported progressive lenticular degeneration as a noticeable clinical entity, when he described four patients with this familial but not hereditary disease. Since then, significant progress has been made to our understanding of the disease (12).
The global prevalence of Wilson’s disease is figured as 12–30 patients per million populations, although clinical profile in terms of varying presentations is presumed to be under-publicized, and in Iran, its accurate prevalence is still not known (13).
In 1929, Vogt et al. (13) explained copper overload as the cause of the disease. In 1985, “Wilson gene” was mapped, and in 1993, ATP7B gene was cloned. Deficit of ATP7B decreases the secretion of copper in bile. Toxic accumulation of excess copper either as free or in bounded form damages organs. Despite variability in phenotypic expression, the disease progresses insidiously and is perpetual with wide clinical spectrum (12).
Generally, the first symptoms are hepatic in 40–45% and neurological in 45% of patients. Renal or osteomuscular findings as initial presentation are rarely reported.
The major difficulty in establishing the diagnosis of Wilson’s disease is it’s infrequent but varied manifestations. Its timely diagnosis is crucial for successful treatment.
Although musculoskeletal features may be well-developed in Wilson’s disease, this has limited consideration. Accepted musculoskeletal manifestations of Wilson’s disease include the following: bone demineralization (14), genu varum (15), scoliosis (16), synovitis (17), osteochondritis dissecans (18), osteomalacia and rickets (19, 20), avascular necrosis of femoral head (21), early osteoarthritis (17), spontaneous fracture (21–23), heterotopic ossification (24), osteoporosis (25), and epiphyseal dysplasia (26).
Osteomuscular aspect is one of the four key criteria of set domains proposed by Global Assessment Scale for Wilson’s disease in 2009 (27). Yu et al., in 2017, came out with isolated musculoskeletal (osseomuscular) type of Wilson’s disease (15).
The diagnosis of Wilson’s disease is based on the results of several clinical and biochemical examinations (28–30). Each of these diagnostic tests has its limitations, and only a combination of clinical, biochemical, and genetic examinations creates a logical criteria for correct diagnosis of Wilson’s disease.
Unexpectedly, the patient presented with proteinuria. Despite having osteopaenia on plain radiograph, he had normal serum vitamin D levels. Owing to abnormal liver function tests and cirrhosis on sonography along with the presence of K-F rings on slit lamp examination, Wilson’s disease was suspected. Nevertheless, low ceruloplasmin and serum copper levels, and high 24-h urinary copper output established the diagnosis. In this case, we did not perform liver biopsy.
In patients with Wilson’s disease, renal involvement in the form of proximal RTA is reported.
Here, RTA is signified by the presence of normal anion gap hyperchloremic metabolic acidosis, and depending on the type of RTA, the serum potassium level could be high, normal, or low. Distal and proximal tubular function defects along with decreased glomerular filtration rate and renal plasma flow ameliorated the risk of urolithiasis (31–39). Fanconi’s syndrome (proximal sodium cotransport defect) also has been described in these patients (40).
Furthermore, about 70–80% patients of of Wilson’s disease have been found to have distal RTA, which is proved with ammonium acid load test, Whether they have kidney stones or not. (41, 42). In addition, urolithiasis has been associated with incomplete distal RTA (43).
In spite of having a systemic nonanion gap, hyperchloremic metabolic acidosis in our patient, anion gap metabolic acidosis was normal along with positive urinary anion gap. Urinary pH was also maintained above 6.5 in all urinary analyses.
Glucosuria in the presence of normal blood sugars and hypophosphatemia, hypouricemic in the absence of uricosuria, hypercalciuria, and urinary amino acids were not evaluated. In our patient, based on the above-mentioned biological and radiological investigations, presence of both proximal and distal RTA was arrived at. Notably, because urolithiasis is linked with distal type 1 RTA, it was the most expected cause of stone development. Hypercalciuria has been also described in patients of Wilson’s disease, and urolithiasis exists in about 40% of patients; however, our patient did not have hypercalciuria.
Renal tubular dysfunction is presumed to be due to direct defect to the tubular epithelium because of copper, or increased absorption of calcium because of skeletal or gastrointestinal abnormalities (44).
Osteomalacia is characterized by the remodulation of bone mineralization; in patients of Wilson’s disease, it is a consequence of dRTA urinary calcium loss and prompted acidosis. Even so, osteomalacia induced by RTA is far more frequent with proximal tubular function defects (Fanconi’s syndrome) in comparison to other renal tubular diseases. Proximal RTA rarely occurs as the presenting characterization of Wilson’s disease (45, 46).
Multiple musculoskeletal manifestations have been described in Wilson’s disease. Early onset of osteoarthritis (OA) is the most common musculoskeletal manifestation. Osteoarticular changes associated with Wilson’s disease include osteoporosis, osteomalacia, and degenerative joint diseases, especially of the knees and wrists. Radiography shows bone fragmentation, osteoarthritis, joint capsule calcification, chondrocalcinosis, osteochondritis dissecans (OCD), and subarticular cysts. Some spinal involvements could also be encountered in these patients such as vertebral body squaring, early lumbar osteoarthritis, and osteochondritis. The present patient demonstrated severe bowing deformity of the upper and lower limbs, metaphyseal widening along with marked generalized osteopenia. Degenerative changes in joint, including large osteophytes, patellofemoral fusion, loss of joint space, and deformities, could be observed in the knees. Similar changes are apparent in other large joints. In the wrists, angulation of the carpal bones and squaring in the head of metacarpal bones are discovered. Chest X-ray evidenced severe deformity of the rib cage, inward bowing of the ribs, and marked kyphoscoliosis. Both hemidiaphragms are elevated and limited space was discovered on left of the lungs.
The considered patient had a combined demonstration of proximal and distal tubular dysfunction, a combination of hypophosphatemia, and acidosis; other injuries from the disease could be an aggravating factor.
The leading cause of rickets is alterations in vitamin D, and not responding adequately to its substitutional treatments. It is, therefore, essential to take into consideration the underlying conditions that may cause metabolic bone diseases such as renal tubular dysfunction, leading to disorder of calcium and phosphate reabsorption and synthesis of 1,25-dihydroxyvitamin D3.
Wilson’s disease has a tendency to be misdiagnosed, since its musculoskeletal manifestations are subtle.
A review of Indian patients reported refractory rickets and renal tubular diseases in approximately one-third of the cases of 25 patients with WD (47, 48).
In a meta-analysis limited only to adult patients of Wilson’s disease, the estimated prevalence rate of osteoporosis, osteopenia, and vertebral fracture was 17.6%, 50.0%, and 8.01%, respectively (49).
Rickets has been described infrequently as a presenting feature of Wilson’s disease. Understanding of this entity is essential, as therapy may improve primary condition and renal involvement such as Fanconi’s syndrome due to Wilson’s disease as well as related biological and clinical manifestations and tubular functions (50–52).
The presented patient had been suffering from renal stones for many years; however, since he was never evaluated for the precise etiology of urolithiasis and underlying diseases, this problem remained concealed.
Thus, in the present patient, role of urologists/nephrologists in the early diagnosis and treatment of RTA and underlying diseases, such as Wilson’s disease, in a timely manner, has been highlighted. Unfortunately, late diagnosis, severity of the disease, and adverse effects significantly affected our patient’s life style and physical appearance.
In spite of the rarity of the manifestations of Wilson’s disease, we emphasize that in case of recurrent or resistant urolithiasis, the main etiology must be examined thoroughly. In any patient presenting questionable symptoms, diagnosis of Wilson’s disease must be taken as a top consideration.
REFERENCES
1. Miacheal Levine A. Common bone and mineral disorders of childhood 381-412: Manual of endocrinology and metabolism. 4th ed. Lippincott, Williams and Wilkins; 2009.
2. Rodriguez-Soriano J. Renal tubular acidosis: The clinical entity. J Am Soc Nephrol. 2002;13:2160–70. 10.1097/01.ASN.0000023430.92674.E5
3. Taly AB, Prashanth LK, Sinha S. Wilson’s disease: An Indian perspective. Neurol India. 2009;57:528–40. 10.4103/0028-3886.57789
4. Goyal JA, Kumar N, Rao SS, Shah VB. Wilson’s disease presenting as resistant rickets. Gastroenterol Res. 2011;4:34–5. 10.4021/gr262w
5. Bajpai A, Bardia A, Mantan M, Hari P, Bagga A. Non-azotemic refractory rickets in Indian children. Indian Pediatr. 2005;42:23–30.
6. Kashoor I, Batlle D. Proximal renal tubular acidosis with and without Fanconi syndrome. Kidney Res Clin Pract. 2019 Sep 30;38(3):267–281. 10.23876/j.krcp.19.056
7. Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003 Jun;23(3):139-42. 10.1034/j.1600-0676.2003.00824.x
8. Wiebers DO, Wilson DM, McLeod RA, Goldstein NP. Renal stones in Wilson’s disease. Am J Med. 1979 Aug;67(2):249–54. 10.1016/0002-9343(79)90399-1. PMid: 463930.
9. Kalra V, Mahajan S, Kesarwani PK. Rare presentation of Wilson’s disease: A case report. Int Urol Nephrol. 2004;36(2):289–91.10.1023/B:UROL.0000034630.73124.36
10. Dziezfiyc-Jaworska K, Litwin T, Członkowska A. Clinical manifestations of Wilson’s disease in organs other than the liver and brain. Ann Transl Med. 2019 Apr;7(Suppl 2):S62. 10.21037/atm.2019.03.30. PMid: 31179299; PMCID: PMC6531658.
11. Wilson SAK. Progressive lenticular degeneration: A familial nervous disease associated with cirrhosis of the liver. Brain. 1912;34(4):295–507. 10.1093/brain/34.4.295.
12. Sandahl TD, Laursen TL, Munk DE, Vilstrup H, Weiss KH, Ott P. The prevalence of Wilson’s disease. An update. Hepatology. 2020;71:722–32. 10.1002/hep.30911.
13. Vogt A. Kupfer und silber aufgespeichert in auge, leber, milz und nieren als symptoms der pseudosklerose. Klin Mbl Augenheilk 1929;83:417–419.
14. Weiss KH, Van de Moortele M, Gotthardt DN, Pfeiffenberger J, Seeßle J, Ullrich E, et al. Bone demineralisation in a large cohort of Wilson’s disease patients. J Inherit Metab Dis. 2015;38:949–956. 10.1007/s10545-015-9815-y
15. Yu H, Xie JJ, Chen YC, Dong QY, Dong Y, Ni W, et al. Clinical features and outcome in patients with osseomuscular type of Wilson’s disease. BMC Neurol. 2017;17:34. 10.1186/s12883-017-0818-1
16. Li Z, Yu X, Shen J, Liang J. Congential scoliosis in Wilson’s disease: Case report and review of the literature. BMC Surg. 2014;14:71. 10.1186/1471-2482-14-71
17. Aslan B, Erdoğan H, Yazısız V. The view of joints in the Wilson’s disease. Eur J Rheumatol. 2020 Apr 28;7(4):205–6. 10.5152/eurjrheum.2020.19184. Epub ahead of print. PMid: 32384047; PMCID: PMC7574760.
18. Merle U, Schaefer M, Ferenci P,Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: A cohort study. Gut. 2007;56:115–120. 10.1136/gut.2005.087262
19. Selvan C, Thukral A, Chakraborthy PP, Rana B, Ajitesh R, Soumik G, et al. Refractory rickets due to Fanconi’s Syndrome secondary to Wilson’s disease. Indian J Endocrinol Metab. 2012;16(Suppl 2):S399–401. 10.4103/2230-8210.104107
20. Goyal JP, Kumar N, Rao SS, Shahl VB. Wilson’s disease presenting as resistant rickets. Gastroenterology Res. 2011;4:34–5. 10.4021/gr262w
21. Verma R, Junewar V, Sahu R. Pathological fractures as an initial presentation of Wilson’s disease. BMJ Case Rep 2013; 2013: pii: bcr2013008857. 10.1136/bcr-2013-008857. PMid: 23576656.
22. Tsuchiya M, Takaki R, Kobayashi F, Nagasaka T, Shindo K, Takiyama Y. Multiple pseudofractures due to Fanconi’s syndrome associated with Wilson’s disease. Rinsho Shinkeigaku. 2017 Sep 30;57(9):527–30. Japanese. 10.5692/clinicalneurol.cn-000953. Epub 2017 Aug 31. PMid: 28855492.
23. Bhatnagar N, Lingaiah P, Lodhi JS, Karkhur Y. Pathological fracture of femoral neck leading to a diagnosis of Wilson’s Disease: A case report and review of literature. J Bone Metab. 2017 May;24(2):135–9. 10.11005/jbm.2017.24.2.135. Epub 2017 May 31.
24. Rodríguez Nieva N, Febrer Rotger A, Meléndez Plumed M, Vernet Bori A. Osteoarthropathy in three siblings with Wilson’s disease. Ann Pediatr (Barc). 2004;61:181–4. 10.1016/S1695-4033(04)78379-4
25. Quemeneur AS, Trocello JM, Ea HK, Ostertag A, Leyendecker A, Duclos-Vallée JC, et al. Bone status and fractures in 85 adults with Wilson’s disease. Osteoporos Int. 2014;25:2573–80. 10.1007/s00198-014-2806-2
26. Park NH, Kim HS, Yi SY, Min BC. Multiple osteochondritis dissecans of knee joint in a patient with Wilson’s disease, focusing on magnetic resonance findings. Knee Surg Relat Res. 2013;25:225–9. 10.5792/ksrr.2013.25.4.225
27. Aggarwal A, Aggarwal N, Nagral A , Jankharia G, Bhatt M. A novel Global Assessment Scale for Wilson’s disease (GAS for WD). Mov Disord. 2009;24:509–18. 10.1002/mds.22231
28. Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: A cohort study. Gut. 2007 Jan;56(1):115–20. 10.1136/gut.2005.087262. Epub 2006 May 18. PMid: 16709660; PMCID: PMC1856673.
29. Schilsky ML. Wilson disease: genetic basis of copper toxicity and natural history. Semin Liver Dis. 1996 Feb;16(1):83–95. 10.1055/s-2007-1007221.
30. Brewer GJ. Recognition, diagnosis, and management of Wilson’s disease. Proc Soc Exp Biol Med. 2000;22:339–46.
31. Morgan HG, Stewart WK, Lowe KG, Stowers JM, Johnstone JH. Wilson’s disease and the Fanconi syndrome. Q J Med. 1962;31:361–84.
32. Mehta RS, Shinde VA. Wilson’s disease with rickets. Neurol India. 1965;13:67–73.
33. Kabra SK, Bagga A, Malkani I. Wilson’s disease presenting with refractory rickets. Indian Pediatr. 1990;27:395–7.
34. Goyal JP, Kumar N, Rao SS, Shah VB. Wilson’s disease presenting as resistant rickets. Gastroenterol Res. 2011;4:34–5. 10.4021/gr262w
35. Joshua GE. Hepatolenticular degeneration (Wilson’s disease) and rickets in children. Indian J Med Res. 1973;61:1876–84.
36. Basu M. Wilson’s disease presenting as a case of resistant rickets. Sri Lanka J Child Health. 2014;43:106–7. 10.4038/sljch.v43i2.7014
37. Ghosh JB, Chakrabarty S, Singh AK, Gupta D. Wilson’s disease—Unusual features. Indian J Pediatr. 2004;71:937–8. 10.1007/BF02830841
38. Patel B, Sardana V. Wilson’s disease presenting as resistant rickets without Fanconi syndrome. Indian J Case Rep. 2020;6(2):90–2. 10.32677/IJCR.2020.v06.i02.016
39. Palkar AV, Shrivastava MS, Padwal NJ, Padhiyar RN, Moulick N. Renal tubular acidosis due to Wilson’s disease presenting as metabolic bone disease. BMJ Case Rep. 2011 Aug 11;2011:bcr0420114121. 10.1136/bcr.04.2011.4121. PMid: 22688476; PMCID: PMC3158360.
40. Subrahmanyam DK, Vadivelan M, Giridharan S, Balamurugan N. Wilson’s disease–A rare cause of renal tubular acidosis with metabolic bone disease. Indian J Nephrol. 2014;24(3):171–174. 10.4103/0971-4065.132017.
41. Osther PJ, Hansen AB, Røhl HF. Distal renal tubular acidosis in recurrent renal stone formers. Dan Med Bull. 1989 Oct;36(5):492–3. PMid: 2805826
42. Tesar V, Mokrejsová M, Marecek Z, Petrtýl J. Distál nírenální tubul árníacidóza u pacientů s Wilson ovouchorobou [Distal renal tubular acidosis in patients with Wilson’s disease]. Sb Lek. 1991 Oct;93(9–10):315–23. Czech. PMid: 1821465.
43. Fuster DG, Moe OW. Incomplete distal renal tubular acidosis and kidney stones. Adv Chronic Kidney Dis. 2018 Jul;25(4): 366–74. 10.1053/j.ackd.2018.05.007. PMid: 30139463; PMCID: PMC7932558.
44. Ağbas¸ A, Bay ED, Bas¸aran MK, Ifikizceli T, Kayhan GK, Özlük Y. Nephrotic range proteinuria in an adolescent with a diagnosis of Wilson’s disease: Questions. Pediatr Nephrol. 2021 Jul;36(7):2101–2102. 10.1007/s00467-021-04947-7. Epub 2021 Feb 2.
45. Clarke BL, Wynne AG, Wilson DM, Fitzpatrick LA. Osteomalacia associated with adult Fanconi’s syndrome: Clinical and diagnostic features. Clin Endocrinol (Oxf). 1995 Oct; 43(4):479–90. 10.1111/j.1365-2265.1995.tb02621.x. PMid: 7586624.
46. Palkar AV, Shrivastava MS, Padwal NJ, Padhiyar RN, Moulick N. Renal tubular acidosis due to Wilson’s disease presenting as metabolic bone disease. BMJ Case Rep. 2011 Aug 11;2011:bcr0420114121. 10.1136/bcr.04.2011.4121. PMid: 22688476; PMCID: PMC3158360.
47. Shin JJ, Lee JP, Rah JH. Fracture in a young male patient leading to the diagnosis of Wilson’s disease: A case report. J Bone Metab. 2015;22:33–7. 10.11005/jbm.2015.22.1.33
48. Kapoor N, Cherian KE, Sajith KG, Thomas M, Eapen CE, Thomas N, et. al Renal tubular function, bone health and body composition in Wilson’s disease: A cross-sectional study from India. Calcif Tissue Int. 2019;105:459–65. 10.1007/s00223-019-00588-z
49. Chenbhanich J, Thongprayoon C, Atsawarungruangkit A, Phupitakphol T, Cheungpasitporn W. Osteoporosis and bone mineral density in patients with Wilson’s disease: A systematic review and meta-analysis. Osteoporos Int. 2018 Feb;29(2):315–22. 10.1007/s00198-017-4295-6. Epub 2017 Nov 6. PMid: 29110062.
50. Yu H, Xie JJ, Chen YC, Dong QY, Dong Y, Ni W, et al. Clinical features and outcome in patients with osseomuscular type of Wilson’s disease. BMC Neurol. 2017 Feb 17;17(1):34. 10.1186/s12883-017-0818-1. PMid: 28212618; PMCID: PMC5316220.
51. Kumar T, Thakar A. Ayurvedic approach for management of Wilson’s disease: A case report. J Ayurveda Integr Med. 2020 Apr–Jun;11(2):177–80. 10.1016/j.jaim.2019.09.004. Epub 2020 Feb 7. PMid: 32044225; PMCID: PMC7329710.
52. Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: A cohort study. Gut. 2007 Jan;56(1):115–20. 10.1136/gut.2005.087262. Epub 2006 May 18. PMid: 16709660; PMCID: PMC1856673.