
Download
CASE REPORTS: NEPHROLOGY
IgA Nephropathy Associated with IgA Vasculitis Coexisting with Focal Segmental Glomerulosclerosis Tip Variant: A Case Report
Zitlali Guadalupe Paulín Zepeda1*, Karla Daniela Salgado Guizar2, Vianey Guadalupe Téllez Bolaños2, Louis Fernando Robles Fernandes2, María Inés Gil Arredondo1
1Department of Nephrology, Centro Médico Nacional Siglo XXI of the Mexican Institute of Social Security, Mexico City, Mexico;
2Department of Internal Medicine resident at Centro Médico Nacional Siglo XXI, Mexican Social Security Institute, Mexico City, Mexico
Abstract
IgA vasculitis (IgAV), previously known as Henoch–Schönlein purpura, is a form of small vessel vasculitis that affects the skin, joints, intestines, and kidneys. The clinical presentation in adults is usually infrequent, more severe, with a worse prognosis and a higher recurrence rate. Due to limited scientific evidence, IgAV is viewed similarly to IgA nephropathy (IgAN), and the renal histopathological lesions are indistinguishable between the two. IgAN is the most prevalent glomerular diseases worldwide. The diagnosis of IgAN is confirmed through a renal biopsy. The most frequently found histopathological lesions are mesangial proliferation and concurrent IgA deposition confirmed by direct immunofluorescence. Focal segmental glomerulosclerosis (FSGS) appears as a glomerular pattern of injury in up to 40% of renal biopsies with IgAN, although there are few reported cases in the international literature where both diseases coexist as primary etiologies. We report a case of a female patient presenting with vasculitic lesions, abdominal pain, and nephrotic syndrome, whose cause is confirmed by a renal biopsy with a diagnosis of IgAN secondary to IgAV and coexistence of FSGS, an unusual presentation with few case reports.
Key words: glomerulosclerosis, IgA nephropathy, IgA vasculitis, nephrotic syndrome
Received: 31 May 2023; Accepted after Revision: 4 July 2023; Published: 14 July 2023
Author for correspondence: Zitlali Guadalupe Paulín Zepeda. Resident Nephrologist, Centro Médico Nacional Siglo XXI of the Mexican Institute of Social Security, Mexico City, Mexico. Email: zitlali.paulin@gmail.com
How to cite: Paulín-Zepeda ZG, et al. IgA Nephropathy Associated with IgA Vasculitis Coexisting with Focal Segmental Glomerulosclerosis Tip Variant. A Case Report J Ren Hepat Disord. 2023;7(2): 10–14.
DOI: 10.15586/jrenhep.v7i2.166
Copyright: Paulín-Zepeda ZG, et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0
Introduction
IgA vasculitis (IgAV) is an extremely rare disease in adults, with an incidence of 0.1–0.8 per 100,000 individuals. The typical clinical presentation includes palpable purpura in 96–100% of cases, joint manifestations in 61%, and gastrointestinal symptoms in 48–53%, usually developing 1 week after the rash. Abdominal pain is constant, localized, and often accompanied by serious complications such as intussusception, infarction, and intestinal perforation (1). Renal involvement occurs in up to 85% of cases with morphologically indistinguishable lesions from IgA nephropathy (IgAN), an autoimmune disease that is the leading cause of primary glomerulonephritis worldwide, with a progression rate of 30–40% to chronic kidney disease (2). The incidence peaks between the second and third decades of life, with a male-to-female ratio of 3:1. In comparison to pediatric cases, the clinical presentation in adults is usually more severe, with a worse prognosis and a higher recurrence rate of the disease after treatment initiation (1).
The variability in worldwide prevalence can be explained by differences in healthcare systems and accessibility to renal biopsies (2). According to the pathophysiology of the disease, there is destruction of the glomerular basement membrane mediated by deposition of immunocomplexes formed by aberrantly degalactosylated IgA1 directed against IgG and C3, resulting in the clinical presence of hematuria, usually subnephrotic proteinuria, hypertension, and less frequently (5–10%) nephrotic syndrome and rapidly progressive glomerulonephritis syndrome (2). Similarly, it has been identified that IgAN presents some morphologically identical lesions to focal segmental glomerulosclerosis (FSGS) as a histological pattern, which may be the result of post-inflammatory scarring, compensatory hemodynamic changes due to loss of nephrons, and primary podocyte damage by concomitant cytokines to the aberrant deposition of IgA1, mainly in the mesangium.
In a study by Khalil et al., it was found that IgAN is associated with this histological pattern but with lesions similar to those of primary FSGS. When these overlap with other glomerular lesions (e.g., mesangial hyperplasia or endocapillary hypercellularity) typical of IgAN, the prognosis worsens (3). The diagnosis of IgAN is confirmed by renal biopsy, which accounts for 40% of reported biopsies in Asia, 20% in Europe, 10% in the United States, and 7% in Mexico (4). The case presented here involves a young female patient who presented with vasculitis skin lesions, acute abdominal pain, normocomplementemic nephrotic syndrome, hypertension, and no impairment of renal function, following an upper respiratory tract infection. A percutaneous renal biopsy was performed, which revealed IgAN along with focal segmental glomerulosclerosis tip variant. The coexistence of both entities as primary diseases is rare in the medical literature, and our case is the first reported in an adult in Mexico.
Case Description
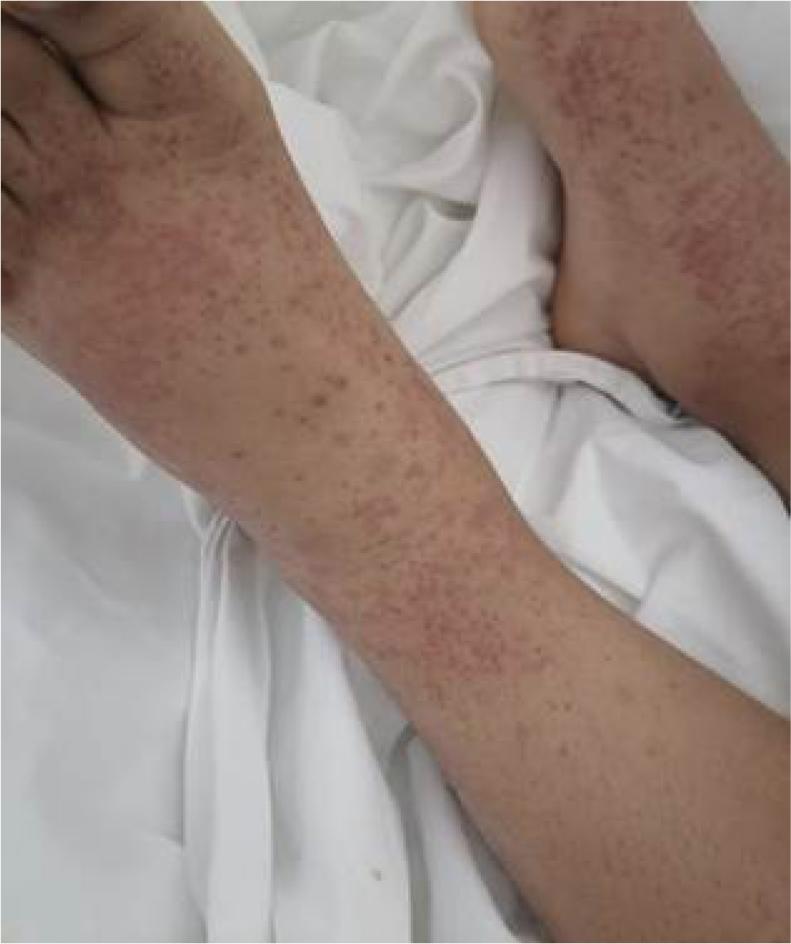
We present the case of a 29-year-old female, with a history of acute myeloid leukemia in 2012, with complete remission 4 years prior to her current condition. Initially admitted to a secondary-care level hospital due to progressive and ascending edema of the pelvic limbs (+++), accompanied by dermatological lesions of papular type that do not disappear upon pressure, confluent and ascending until becoming generalized (Figure 1), which appeared after a flu-like illness. She also had arterial hypertension with hypertensive emergency (185/100 mmHg), abdominal pain with a visual analogue scale rating of 9/10, leading to exploratory laparotomy, which was reported as normal. Subsequently, she was evaluated by dermatology at the same facility, and a skin biopsy was performed, revealing non-granulomatous neutrophilic dermatitis. In her initial laboratory tests, notable findings included the following: glucose 149 mg/dL, urea 49 mg/dL, ureic nitrogen 23 mg/dL, serum creatinine 0.63 mg/dL, albumin 3.4 mg/dL, total proteins 5.4 mg/dL, globulins 1.9 mg/dL, calcium 8.5 mg/dL, sodium 135 mEq/L, chloride 105 mEq/L, potassium 3.9 mEq/L, phosphorus 3.4 mg/dL, magnesium 1.6 mg/dL, and a 24-h urine collection showing proteinuria of 9 g. She was referred to our unit, where further diagnostic workup was conducted, including immunological tests showing normal complement levels, negative C-ANCA and P-ANCA, negative lupus anticoagulant, negative anticardiolipin antibodies, negative anti-dsDNA, negative ANAs, negative anti-Ro and anti-La antibodies, protein electrophoresis without evidence of monoclonal pattern, and a subcutaneous periumbilical tissue biopsy with negative Congo red staining for amyloid deposits.
Figure 1: Maculopapular dermatosis observed on patient’s pelvic limbs. By morphology, they were suggestive of vasculitis.
Considering the reported medical history, a deliberate search for secondary causes of nephrotic syndrome was conducted, ruling out rheumatologic diseases, amyloidosis, and multiple myeloma. Hematologic neoplastic activity was also intentionally ruled out due to the history of acute myeloid leukemia through peripheral blood smear, which showed no oncological pathology.
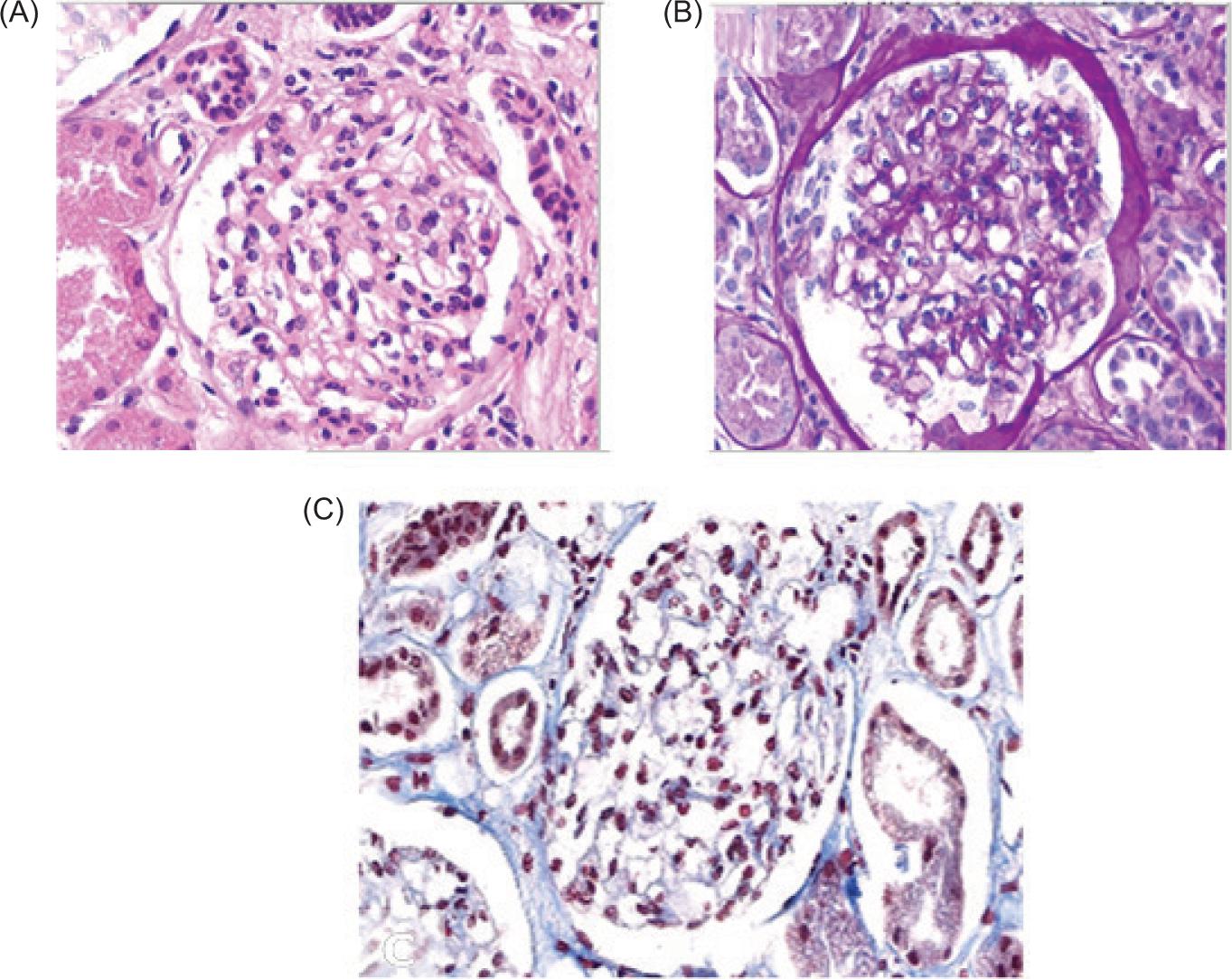
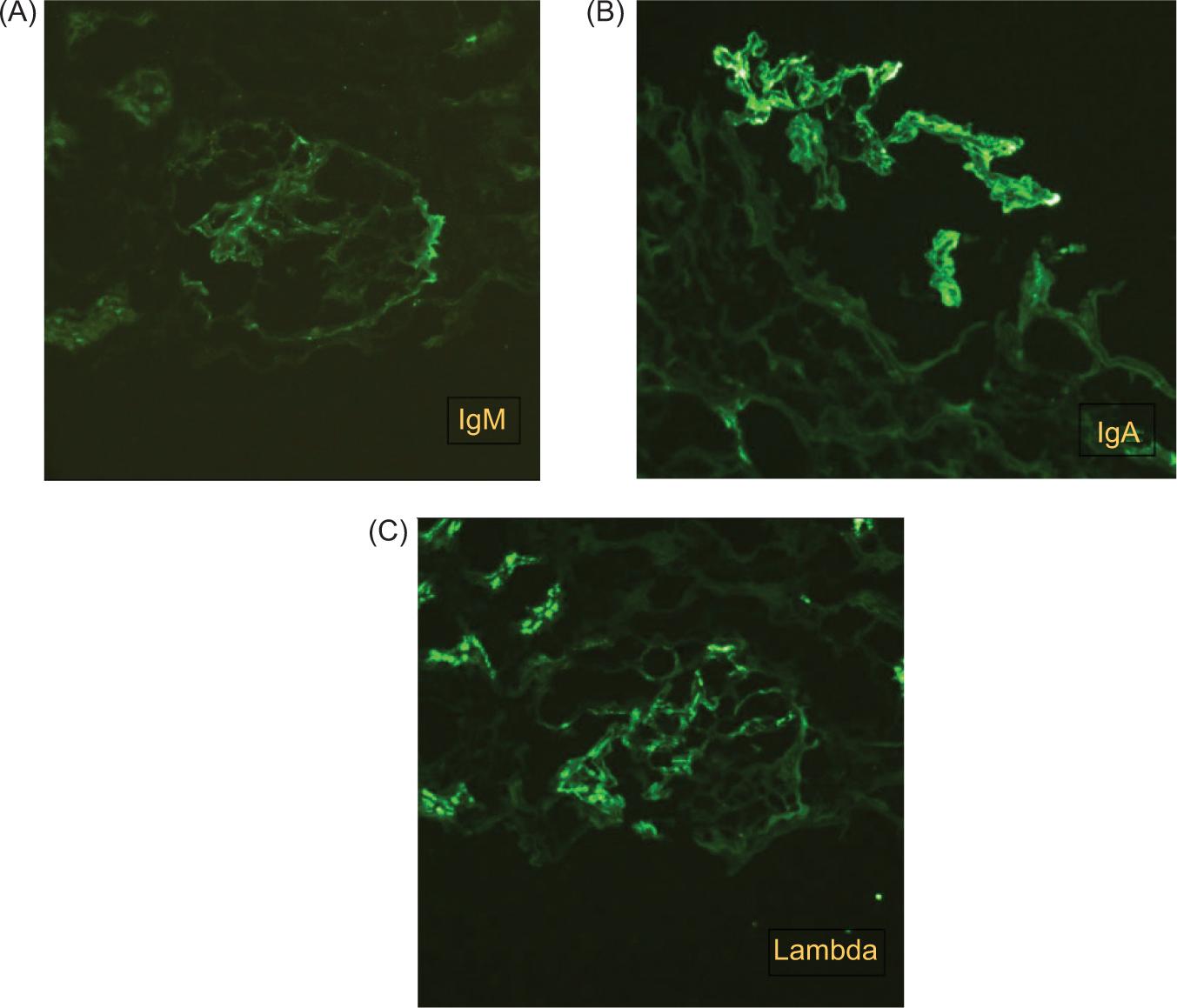
During the patient’s hospital surveillance, infectious foci were also ruled out through blood and urine cultures, which yielded negative results. A follow-up chest X-ray was performed, showing no pleuropulmonary abnormalities. A renal biopsy was carried out (Figure 2), which reported IgAN (M1, E1, S1, T0, C1) with FSGS tip variant, focal acute tubular injury with mild epithelial regeneration, and Grade 1 interstitial fibrosis. Additionally, direct immunofluorescence was performed, demonstrating dominant deposits of IgA (Figure 3).
Figure 2: Renal cortex biopsy with a total of 15 glomeruli. (A) Hematoxylin and Eosin staining: Glomerulus showing global sclerosis, as well as segmental sclerosing lesion of a scar-like type, forming adhesions between the capillary tuft and the Bowman’s capsule at the tubular pole. (B) Periodic Acid-Schiff (PAS) staining: Glomerulus with diffuse expansion of the mesangial matrix, along with folded and scarred segments. (C) Masson’s trichrome staining: Interstitium displaying areas of fibrosis and associated tubular atrophy affecting approximately 10–15% of the cortical surface.
Figure 3: Direct immunofluorescence. (A) IgM: Positive with a focal and segmental granular pattern in the mesangium (1+). (B) IgA: Positive with a global and diffuse granular pattern in the mesangium (3+). (C) Lambda light chain: Positive with a global and diffuse granular pattern in the mesangium (3+).
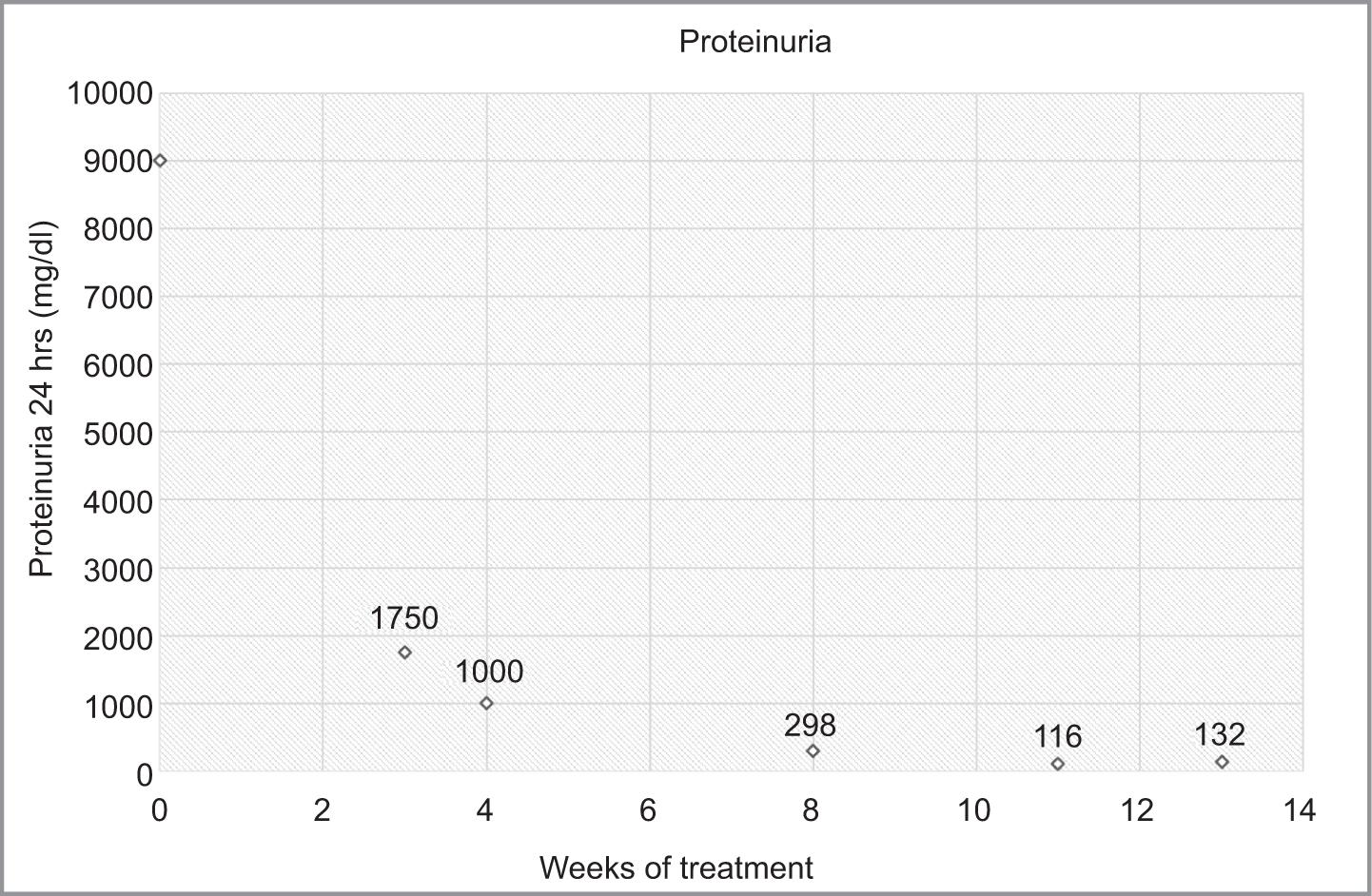
It is concluded that the patient had IgAN associated with FSGS of the tip variety. After the renal biopsy and improvement of general conditions, it was decided to discharge the patient with a treatment plan taking into account the clinical presentation, preserved renal function, and absence of life-threatening extrarenal manifestations of vasculitis such as alveolar hemorrhage. The treatment plan included antiproteinuric and antihypertensive management targeted at IgAN, as well as prednisone at a dosage of 1 mg/kg/day targeting FSGS, according to the KDIGO 2021 guidelines for glomerular diseases. A gradual reduction in the prednisone dosage was indicated starting from the fourth week of treatment. Follow-up was offered through outpatient nephrology consultations to monitor proteinuria and blood pressure, including a proteinuria curve (Figure 4) that showed a decreasing trend and achieved complete remission within the therapeutic range at a 16-week regimen, as recommended by international therapeutic guidelines.
Figure 4: Follow-up graph of proteinuria in the patient from diagnosis until week 14, within a 16-week regimen to evaluate the response to treatment.
Discussion
The presence of IgAV associated with IgAN and FSGS as primary entities is extremely rare, especially in adult patients, where the prognosis is usually grim. There is a predominance in male patients, with an average age of 58 years (±18) (5, 6). Clinically, palpable purpura is the predominant characteristic, followed by renal manifestations, which are similar to those seen in IgAN. The indications for renal biopsy in cases of IgAV include clinical data of IgAN, such as proteinuria >1 g/day, deterioration of renal function, or rapidly progressive syndrome (7).
From the histopathological point of view, there are no differences between the lesions of one entity and the other. The treatment depends on the clinical spectrum of the patient and may range from supportive management including lifestyle modifications, antiproteinuric measures, and antihypertensive drugs to an immunosuppressive regimen in selected cases.
We present a female patient in her third decade of life who presented with pharyngitis-like symptoms and palpable purpura in the lower extremities. Although this raised suspicion of IgAV, the appearance of edema and proteinuria of 9 g in 24 h opened up the possibility of other differential diagnoses, as nephrotic syndrome is a rare presentation in IgAV and IgAN (10%).
Secondary etiologies that could justify the occurrence of nephrotic syndrome were intentionally ruled out, such as viral infections, neoplasms, amyloidosis, monoclonal gammopathies, and autoimmune diseases, particularly systemic lupus erythematosus, given that the patient belonged to an age group highly associated with this entity. It is well known that the abnormal galactosylation present in the hinge region of IgA is responsible for the aberrant formation of IgA, which, when combined with IgG and C3, forms immune complexes that damage the glomerular basement membrane, causing direct podocyte injury due to inflammatory mediators and subsequent deposition in the mesangium (8). This mechanism could explain the presentation of nephrotic syndrome in IgAN. However, upon obtaining the results of the renal biopsy, focal and segmental glomerular lesions were found, with a histological pattern of tip variant and mesangial IgA deposition in direct immunofluorescence. Regarding prognosis, the tip variant of focal segmental glomerulosclerosis has a better renal function prognosis, with a low progression rate to chronicity of 5–20% (9).
Conclusions
Considering that the patient maintained preserved renal function throughout her evolution and had no life-threatening extrarenal manifestations such as alveolar hemorrhage, it was decided to initiate supportive management with antiproteinuric and antihypertensive measures, along with targeted treatment for the tip variant of FSGS using prednisone at a dose of 1 mg/kg/day. The patient achieved partial remission of proteinuria at 3 weeks and complete remission at 8 weeks after the initiation of treatment, indicating a good response. Currently, 1 year after the start of treatment, the patient continues to have complete remission of proteinuria and no deterioration in renal function, which confers a favorable prognosis regarding the progression of the disease toward chronicity. This clinical case is the first reported association of IgAN and FSGS in an adult with description of treatment success in Mexico. Although rare, its prognosis became favorable and with good treatment response.
The authors declare no potential conflicts of interest with respect to research, authorship, and/or publication of this article.
Declaration of Consent
Informed consent was obtained from the patient authorizing the development of this case report.
REFERENCES
1. Maritati F, Canzian A, Fenaroli P, Vaglio A. Adult-onset IgA vasculitis (Henoch-Schönlein): Update on therapy. Presse Med [Internet]. 2020;49(3):104035. 10.1016/j.lpm.2020.104035
2. Rajasekaran A, Julian BA, Rizk DV. IgA nephropathy: An interesting autoimmune kidney disease. Am J Med Sci [Internet]. 2021;361(2):176–94. 10.1016/j.amjms.2020.10.003
3. El Karoui K, Hill GS, Karras A, Moulonguet L, Caudwell V, Loupy A, et al. Focal segmental glomerulosclerosis plays a major role in the progression of IgA nephropathy. II. Light microscopic and clinical studies. Kidney Int. 2011;79(6):643–54. 10.1038/ki.2010.460
4. Chavez V, Orizaga C, Becerra J, Fuentes F, Parra R, Aragaki Y, et al. Epidemiología de la enfermedad glomerular en adultos. Revisión de una base de datos [Internet]. 1ª ed. 2014 [citado
5. Rodrigues JC, Haas M, Reich HN. IgA ephropathy. Clin J Am Soc Nephrol. 2017;12(4):677–86. 10.2215/cjn.07420716
6. Pillebout E. Henoch–Schonlein purpura in adults: Outcome and prognostic factors. J Am Soc Nephrol. 2002;13(5):1271–8. 10.1097/01.asn.0000013883.99976.22
7. Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 clinical practice guideline for the management of glomerular diseases. Kidney Int. 2021;100(4S):S1–S276. 10.1016/j.kint.2021.05.021
8. Rawla P, Limaiem F, Hashmi MF. IgA nephropathy. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023.
9. Shabaka A, Tato Ribera A, Fernández-Juárez G. Focal segmental glomerulosclerosis: State-of-the-art and clinical perspective. Nephron [Internet]. 2020;144(9):413–27. 10.1159/000508099