
Download
REVIEW: HEPATOLOGY
The Interplay of Iron and Lipid Homeostasis in Non-Alcoholic Fatty Liver Disease
Clinton J. Kidman1,2,3, Cyril D.S. Mamotte1,2, Keea R. Inder-Smith1,2, Mark J. Tobin4, Mark J. Hackett2,5, Ross M. Graham1,2*
1School of Medicine, Curtin University, Perth, Western Australia;
2Curtin Health Innovation Research Institute, Curtin University, Perth, Western Australia;
3Department of Chemistry, University of Adelaide, Adelaide, South Australia;
4ANSTO – Australian Synchrotron, Clayton, Victoria, Australia;
5School of Molecular and Life Sciences, Curtin University, Perth, Western Australia
Abstract
The liver is essential for numerous metabolic functions and is the primary site of iron storage and regulation in addition to maintaining critical functions in lipid metabolism. Both iron deficiency and overload have been demonstrated as being involved with metabolic dysfunction; hence, tight regulation of iron absorption is essential to maintain health. Approximately one-third of individuals suffering from non-alcoholic fatty liver disease have elevated hepatic iron concentrations, with increased iron associated with increased disease severity, suggesting a convergence in dysregulation between lipid and iron metabolism. Increasingly, the literature is demonstrating, using a myriad of model organisms and iron-loading methods, that iron loading induces dysregulation in multiple aspects of hepatic lipid metabolism. However, the molecular mechanisms involved, and their subsequent effects on human diseases, are unclear. As iron is a fundamental component of many enzymes and proteins involved in lipid metabolism and is involved in the production of free radicals and oxidative stress, the mechanisms are numerous. In this review, we examine and summarise the dysregulation that iron loading elicits on hepatic lipid availability, de novo synthesis, catabolism, and export. We propose that understanding the interplay between iron and lipid metabolism holds the key to unlocking the complexities of disease development and progression, ultimately leading to improved therapeutic avenues.
Key words: cholesterol, iron metabolism, lipid metabolism, non-alcoholic fatty liver disease, triglycerides
Received: 4 July 2023. Accepted after revision: 18 December 2023. Published: 20 April 2024
Authors for correspondence: Ross M. Graham, and Clinton J. Kidman, Curtin Health Innovation Research Institute, Building 305, Curtin University, Bentley 6102, Western Australia. Emails: rmgraham@curtin.edu.au; clinton.kidman@adelaide.edu.au
How to cite: Kidman C.J., et al. The Interplay of Iron and Lipid Homeostasis in Non-Alcoholic Fatty Liver Disease. J Ren Hepat Disord. 2024;8(1): 1–16.
DOI: 10.15586/jrenhep.v8i1.170
Copyright: Kidman C.J., et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0
Introduction
Iron is an essential trace element and is a critical structural and functional component of many physiological systems. It exists in multiple forms in mammals, as labile iron, or as complexed in transferrin, ferritin, haemosiderin and haem-containing proteins, such as haemoglobin, myoglobin and haemo-enzymes (1, 2). Iron facilitates electron transfer (3), allowing it to catalyse enzymatic reactions and mediate oxygen transport, mitochondrial respiration, and DNA biosynthesis (4). However, unregulated electron transfer may result in production of reactive oxygen and nitrogen species causing oxidative stress and cellular damage (5). The toxicity of iron is mitigated by regulation of iron metabolism, which starts with tight control of intestinal absorption (6). The average adult body contains 3–5 g of iron, with only 1–2 mg of iron being absorbed daily by the duodenum under healthy conditions so as to replace iron lost through mechanisms such as sweating, desquamation and menstruation (7). There is no controlled mechanism to eliminate excess iron once absorbed.
The liver is central to regulating whole body iron homeostasis, being the major location of iron storage and the production of the master iron regulating hormone, hepcidin (8, 9). Despite being tightly regulated, iron dysregulation, leading to deficiency or accumulation, can occur as the result of genetic (10) or nutritional factors (11). Mild to moderate iron accumulation is becoming increasingly frequent in the general population and is particularly associated with metabolic syndrome and its hepatic manifestation, non-alcoholic fatty liver disease (NAFLD), which has a global prevalence of approximately 30% (12–16).
The liver also plays a key role in maintaining lipid homeostasis and is a hub for fatty acid (FA) synthesis and circulating lipids through lipoprotein synthesis (17, 18). Under healthy conditions, a small fraction of the large quantity of fatty acids processed by the liver is stored within hepatocytes in the form of triglycerides (TG) (19, 20). Dysregulation of lipid metabolism is increasingly common among the global population (21, 22), leading to increased prevalence of hepatic lipid droplets and, eventually, NAFLD (23). The liver therefore represents a nexus for interaction between iron and lipid metabolic pathways.
Iron metabolism
Iron is a critical structural and functional component of multiple physiological systems, and these systems utilise iron’s ability to switch between oxidation states to mediate electron transfer (24). However, uncontrolled electron transfer is toxic through the generation of free radicals, which can, in turn, induce oxidative stress (5). As mammals, including humans, lack the ability to excrete iron in a controlled manner, control of body iron is achieved through regulation of iron absorption. The liver plays a fundamental role in the regulation of iron, being the major site of iron storage and the production of hepcidin, a peptide hormone responsible for inhibiting iron’s entry into the plasma from enterocytes (9).
Hepatocytes account for ~98% of hepatic iron storage under typical conditions (25). Most of the iron (~80%) is bound to ferritin, which acts as an iron reserve to maintain iron availability and to protect against oxidative damage. Transferrin and haem account for the majority of the remaining iron, ~5% and ~2–3%, respectively. Intracellular iron not specifically bound to proteins or other high molecular weight molecules is referred to as the labile iron pool, an ill-defined pool of chelatable and potentially redox-active iron bound to low molecular weight chelators (26). Despite the multiple levels of regulation, dysregulation of iron is associated with diseases such as diabetes and NAFLD (15, 27).
Iron in the context of NAFLD
Iron accumulation is inextricably linked with NAFLD, with ~30% of NAFLD individuals presenting with mild to moderate hepatic iron loading (28). The role of iron in NAFLD progression has been extensively studied through the lens of increased oxidative stress, which is a major contributing factor in the development of non-alcoholic steatohepatitis (NASH). Iron causes oxidative stress by catalysing the formation of free radicals (5), and markers of oxidative stress are known to increase with hepatic iron accumulation (29, 30). In NAFLD, oxidative stress can lead to depletion of adenosine triphosphate (ATP), oxidised nicotinamide adenine dinucleotide (NAD+) and glutathione, lipid peroxidation, breaks in DNA strands, and damage to proteins, thereby altering their functions (31–33), leading to cell death.
The clinical significance of increased hepatic iron on progression of NAFLD is slowly becoming clearer, with modest increases in hepatic iron associated with more advanced NAFLD, liver injury and hepatocellular carcinoma (28, 34). However, it is also becoming clear that the role of iron is more complicated than just oxidative stress. Iron has been shown to have a role in altering hepatic equilibrium between lipid uptake, synthesis, oxidation, and export, leading to lipid accumulation due to impairment of mitochondrial β-oxidation gene and protein expression (35, 36), and dysregulated lipid synthesis (37–39). In mice, we demonstrated that iron loading increased the expression of genes involved in the biosynthesis of cholesterol (40) and increased lipid polyunsaturation in a hepatic cell line, potentially increasing susceptibility to oxidative stress (41). This review focuses on the role of iron loading in the dysregulation of lipid metabolism and the consequences for the development and progression of NAFLD.
Plasma lipid availability
Hepatic fatty acid is derived primarily from four sources: lipolysis of adipose tissue, de novo lipogenesis, clearance of chylomicron remnants and dietary lipids (18, 42–44). Under normal conditions, large quantities of fatty acid are processed by the liver. However, the rate of fatty acid acquisition is balanced by the rate of catabolism and secretion, and only a fraction (<5%) is stored within hepatocytes in the form of TG. Isotope studies have shown that the majority of hepatic fatty acids (59%) under NAFLD conditions originate from adipose tissue, compared to 26% from hepatic de novo lipogenesis and 15% from diet (42). In the context of iron loading, it is frequently observed that iron increases plasma free fatty acids (FFA), TG and cholesterol, leading to hyperlipidaemia in rodents and humans (45–48), and suggesting a potentially significant role in the development of NAFLD. To date, iron has been demonstrated to affect mechanisms involved in increasing adipose tissue lipolysis and, thus FFA, and decreasing clearance of TG-rich lipoproteins (46), both of which may result in increased plasma lipids.
In both liver and adipose tissue, the process of hydrolysis of TG to FFA and glycerol requires three enzymes, the first of which, adipose triglyceride lipase (ATGL), also known as patatin-like phospholipase domain-containing protein 2, is the rate-limiting step and catalyses the hydrolysis of TG to diglycerides (DG) (49–51). The resultant DG are further hydrolysed by hormone sensitive lipase (LIPE), producing monoglycerides (MG). The final hydrolysis of MG to glycerol and FFA is catalysed by monoacylglycerol lipase (MGL) (49, 52). Increased expression of the Lipe gene was observed in the epididymal adipose tissue of mice with dietary haem-induced iron loading (53). Consistent with this finding, carbonyl iron-loaded mice have elevated Atgl and Lipe protein with a greater proportion of both phosphorylated in adipose tissue (54). The same mice exhibited increased glycerol and FFA concentrations in subcutaneous and visceral adipose tissue, strongly suggesting an increase in lipolysis (54). Surprisingly, mice injected with iron dextran were shown to have decreased Lipe gene expression with no effect on Atgl gene expression; nevertheless, Atgl protein and the ratio of phosphorylated to non-phosphorylated Lipe increased (55), consistent with observations in dietary iron loading (53, 54). Moreover, in vitro studies demonstrated that iron sulphate and transferrin increased the rate of lipolysis in primary rodent adipocytes in a dose-dependent manner (56, 57). Studies investigating the clinical relevance of iron loading reported an association between iron and lipolysis, with high plasma ferritin levels in obese women associated with high protein levels of both LIPE and ATGL (58). However, it must be recognised that serum ferritin is not a very specific marker of body iron and is also associated with other pathologies, including inflammatory conditions, of which NAFLD is one (59). Additionally, a study of 492 participants over the age of 40 years with a medical connection with type 2 diabetes mellitus showed an increase in fed and fasting plasma FFA concentration with multiple plasma markers of iron loading, including ferritin, transferrin, serum iron and non-transferrin-bound iron (NTBI). NTBI exhibited the strongest association, suggesting that the mechanism may involve lipid peroxidation (48).
The mechanism by which iron loading increases LIPE activity and lipolysis in adipocytes appears to differ between in vitro and in vivo models. In vitro iron loading of adipocytes indicates the mechanism as being independent of protein kinase A (PKA) activation via an unresolved pathway potentially involving lipid peroxidation (56). However, in vivo models suggested that the mechanism involved the upregulation of the β-adrenergic signalling pathway (55) and increased PKA activation (54). Consistent with the proposed mechanism in vivo, iron loading decreased adiponectin gene expression in adipose tissue and reduced plasma levels (60). Adiponectin is known to suppress Lipe activation (61, 62) and its gene expression is inhibited by β-adrenergic signalling via PKA (63).
Lipid uptake and trafficking
Notwithstanding the ability of FFA to diffuse across a lipid bilayer (64), a variety of proteins facilitate FFA transport across cellular membranes, including fatty acid transport proteins (solute carrier protein 27 [SLC27A1-6], previously referred to as FATP1-6), many of which exhibit acyl-CoA synthetase (ACS) activity (65), fatty acid translocase (FAT/CD36) and fatty acid binding proteins (FABPs). The scavenger receptor, CD36, and SLC27A2 and SLC27A5 are responsible for the majority of hepatic fatty acid import (66–68). In vitro models using primary mouse and human hepatocytes showed no change in CD36 gene or protein expression with iron loading (45, 69). These studies, however, demonstrated that iron’s effect on lipid uptake by hepatocytes was modulated by the availability of fatty acids; iron loading with a combination of oleic and palmitic acids (2:1) decreased lipid content and Cd36 protein expression in primary mouse hepatocytes (45). Interestingly, 24-h and 72-h iron-loaded primary human hepatocytes exhibited reduced expression of the SLC27A5 gene, but the subsequent addition of palmitic acid increased both SLC27A5 and CD36 gene expression, compared to control and palmitic-only cells, indicating fatty acid saturation may alter iron’s influence (69). In a model using C. elegans, iron loading increased lipid accumulation in both presence and absence of oleic acid through a mechanism dependent on ACS-20, an orthologue of mammalian SLC27A1/ SLC27A4 (70). In contrast, iron-loaded mouse models exhibited either no effect or reduced gene and protein expression of hepatic Slc27a2, Slc27a5 and Cd36 (71–73). Similarly, primary iron loading induced by hemojuvelin (Hjv) knockout (KO) did not change the hepatic protein expression of Slc27a2 or Slc27a5 (73) and no changes in gene expression were reported (38). Hence, iron loading did not appear to alter the expression of lipid import proteins in mouse models under either genetic (primary) or dietary iron loading. However, similar to in vitro observations, ApoE KO mice fed with a high fat diet with iron loading exhibited reduced Cd36 and Slc27a1 gene expression and Cd36 protein expression, while at the same time demonstrating increased serum FFA and no change in hepatic FFA, suggesting either no change or a reduced hepatic lipid uptake (45). It therefore appeared that lipid availability and composition modulated the effect of iron loading on hepatic lipid import machinery.
The primary function of FABPs is considered as being one of lipid chaperones; however, studies have also demonstrated a role in fatty acid import, storage and export (74). FABP1 is the most abundant FABP in hepatocytes and represents up to 5% of all cytosolic proteins (75, 76). FFAs are known as cytotoxic to cells (77, 78), and their binding to FABPs aids in decreasing their toxicity (79). Additionally, changes in FABP1 expression substantially affect the rate of fatty acid uptake, a property that other FABPs do not demonstrate (80, 81). Interestingly, the effect of iron loading on expression of FABPs is not uniform across all members in both in vitro and in vivo studies. Iron loading in primary mouse hepatocytes was demonstrated not to alter Fabp1 protein expression (45), while, in primary human hepatocytes, iron loading reduced FABP4 in an iron dose-dependent manner but increased FABP5 expression (69, 82). Studies using dietary iron-loaded mice demonstrated reduced hepatic FABP1 protein (73), corroborated in vitro findings of reduced FABP4 and increased FABP5 gene expression, but exhibited no change in FABP2 (71, 82). Although not reaching statistical significance, proteomic studies from iron-loaded mice were consistent with gene expression observations (73). Genetic models of iron loading showed the same relationship with hepatic FABPs as dietary models (73, 82, 83), although Fabp5 gene expression was reduced in Hjv KO mice (82). Hepatic Fabp2 gene and protein expression were similarly shown to be reduced in both Hfe and Hjv KO mouse models (82, 83). These dietary and genetic studies consistently demonstrated that iron loading reduced hepatic FABPs, consistent with reduced uptake and intracellular trafficking of fatty acids.
The introduction of a high lipid environment together with iron loading affects iron’s relationship with FABPs. The addition of palmitic acid plus iron to primary human hepatocytes increased gene expression of both FABP4 and FABP5 (69), while primary mouse hepatocytes loaded with both oleic acid and palmitic acid plus iron exhibited reduced Fabp1 protein (45). Similarly, ApoE KO mice fed with a high iron and high fat diet also exhibited reduced hepatic Fabp1, Fabp2, Fabp4 and Fabp5 gene and protein expression (45). In contrast, Hfe KO mice fed with a high fat diet exhibited increased hepatic Fabp1 gene expression, compared to both wild type and Hfe KO mice fed with a control diet (84). Together, these findings indicated that Fabp expression was sensitive to the composition of high fat treatment and genetic background of the model, demonstrating the importance of considering these factors when designing and interpreting such studies.
The liver is additionally able to import lipids through lipoprotein receptors (85–87). The low-density lipoprotein (LDL) receptor mediates the endocytosis of cholesterol-rich LDL from the plasma and recognises apoprotein B100 (86). Iron-loaded mice demonstrated no change in hepatic low density lipoprotein receptor (LDLR) mRNA or protein expression, suggesting that iron may not alter lipoprotein uptake (36, 46, 88). However, iron loading decreased the activity of lipoprotein lipase (LPL) (46), causing a reduction in clearance of lipoproteins from the plasma by multiple tissues, including adipose tissue, skeletal muscle, and the liver, and suggesting that reduced clearance of lipids could be partially or fully responsible for the elevated plasma lipids observed (89–91).
De novo lipid synthesis
Under normal circumstances, the hepatic de novo lipogenesis (DNL) pathway is rarely utilised (92). Nevertheless, just as enhanced fatty acid uptake can contribute to steatosis and NAFLD development, so does increased DNL (93, 94). DNL is the synthesis of fatty acid chains up to 16 carbons long (palmitate; C16:0) from acetyl-CoA subunits generated though glycolysis and other pathways (49, 95). The first step of DNL is the cleaving of citrate to form acetyl-CoA by ATP-citrate lyase (ACLY) (96, 97), followed by carboxylation of acetyl-CoA to malonyl-CoA by acetyl-CoA carboxylase (ACC/ACACA), the rate limiting step in DNL (98) (Figure 1). The final step in palmitate synthesis of adding acetyl-CoA to malonyl-CoA and subsequent cycles is carried out by fatty acid synthase (FAS) (99). In dietary iron-loaded mice, Acaca and Fas gene and protein expression were upregulated, and iron loading also increased their activity in rats (38, 100, 101). Thus, dietary iron loading upregulated the DNL pathway, leading to increased fatty acid synthesis. These observations were supported by a study reporting that iron loading of primary human umbilical vein endothelial cells (HUVECs) increased FAS gene expression and elevated the rate of 16:0, 16:1 and 18:1 fatty acid synthesis (37). Furthermore, iron loading increased expression of Acc2, the mitochondrial isoform of Acaca (45). Malonyl-CoA produced by Acc2 downregulated mitochondrial β-oxidation by inhibiting Cpt1 (102, 103), indicating iron loading may reduce Cpt1 activity. Although both hepatic ACACA and FAS expression increased with dietary iron loading, Acly gene and protein decreased, suggesting that increases in DNL may utilise a different source of acetyl-CoA (71, 73, 96). Rodent models in which iron loading was induced by bypassing duodenal regulation by injection of iron oxide nanoparticles (IONPs) or iron dextran showed decreased Acly, Acaca and Fas gene and protein expression, demonstrating differences between the two modes of iron loading (104, 105). In contrast, when mice fed with a high fat diet were iron-loaded by either dietary iron or injected IONPs, Acaca and Fas genes and proteins exhibited hepatic upregulation (45, 47).
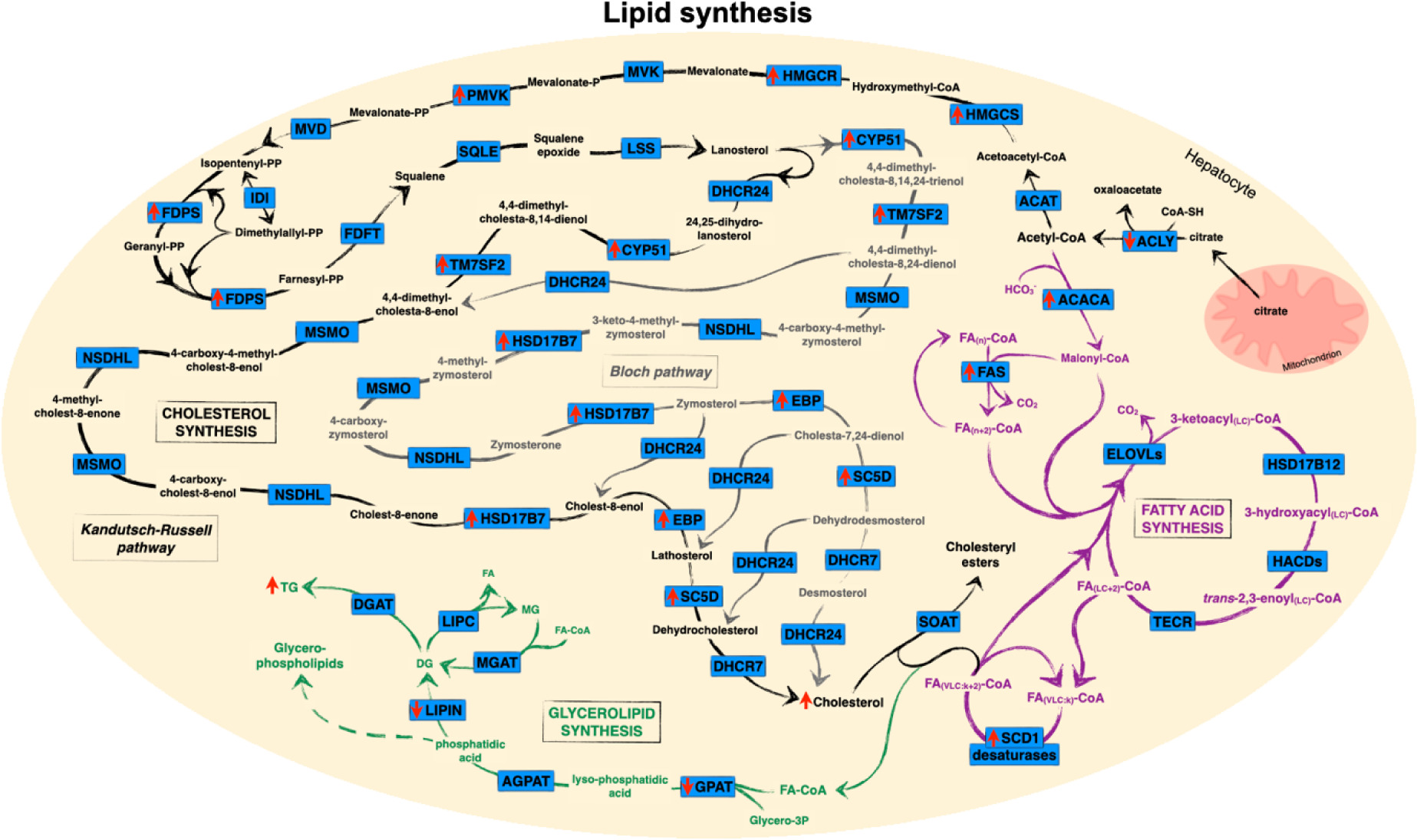
Figure 1 Association between hepatic iron accumulation and dysregulated lipid synthesis. Red arrows indicate direction of iron-associated regulation of gene, protein or metabolite expression as discussed in the text. The Bloch pathway of cholesterol synthesis is shown in black, with the branch to the Kandutsch–Russell pathway and its interconversions with the Bloch pathway shown in grey. Fatty acid synthesis, elongation and desaturation are shown in purple and glycerolipid synthesis is shown in green. In order to simplify the diagram, only some carbon numbers are shown and FA-CoAs destined for incorporation into glycerolipids are shown as being sourced from the pool of very long-chain FAs but may be sourced from any pool of cytosolic FA-CoA. FA(n): fatty acid containing n carbons; FA(LC:k): long-chain (or very long-chain [VLC]) fatty acid containing k double bonds. ATGL, adipose triglyceride lipase; DHCR24, dehydrocholesterol reductase; HACD, hydroxyacyl-CoA dehydratase; LIPC, hepatic lipase; LSS, lanosterol synthase; MGAT, monoglyceride acyltransferase; MSMO, methylsterol monooxygenase; MVD, mevalonate diphosphate decarboxylase; MVK, mevalonate kinase; NSDHL, NAD(P)-dependent steroid dehydrogenase-like; SOAT, sterol O-acyltransferase; SQLE, squalene epoxidase; TECR, trans-2,3-enoyl-CoA reductase. Other abbreviations are defined in the text. Pathways are based on data from KEGG (181) and Mazein et al. (182).
Fatty acid elongation
Fatty acid elongation involves the addition of two-carbon units to a fatty acyl-CoA and is catalysed by four enzymatic reactions (106, 107) (Figure 1). In mammals, the initial step is a rate controlling condensation reaction, catalysed by enzymes referred to as elongation of very long-chain fatty acids (ELOVLs) (106). At present, seven ELOVL proteins have been identified: ELOVL1, ELOVL3, ELOVL6 and ELOVL7 prefer saturated and monounsaturated fatty acids as substrates whereas ELOVL2, ELOVL4 and ELOVL5 are selective for polyunsaturated fatty acids (108–110). ELOVL6 is involved in the elongation of C16:0 to C18:0 and C16:1 to C18:1 (111). Current literature reporting the effect of iron loading on hepatic ELOVL6 is contradictory, with both increased and decreased gene expression observed in C57BL/6 mice fed with similar concentrations of carbonyl iron for similar durations (38, 47, 71). ELOVL2 elongates C20:4 to C22:4 and C20:5 to C22:5 and protein levels were increased in rats fed with a high fat diet plus ferric citrate, although no change in activity was evident (112). Nevertheless, increases in the ELOVL family of proteins were consistent with increases in synthesis of longer-chain fatty acids and may explain the increase in fatty acid chain length observed in AML12 cells incubated with ferric ammonium citrate (41). Elovl3 and Elovl5, which are believed to be involved in the elongation of multiple length fatty acids, exhibited reduced gene expression in mice fed with 2% carbonyl iron (38, 71).
Fatty acid desaturation
Upregulation of hepatic stearoyl CoA desaturase (Scd1) mRNA, protein and enzyme activity in rodents was reported in studies utilising both dietary and injection models of iron loading (47, 113, 114). SCD1 is the first enzyme involved in fatty acid desaturation and introduces a single double bond at ∆9 to produce monounsaturated fatty acids (C16:1 and C18:1) (115) (Figure 1). Scd1 protein increased proportionately with iron status in mice (113). Similarly, hepatic Scd1 mRNA increased in ApoE KO mice fed with a high fat and high iron diet (45), suggesting dietary iron overload increased fatty acid desaturation. Interestingly, Hfe KO mice did not demonstrate altered hepatic Scd1 gene expression compared to wild-type mice (116), potentially indicating a difference of iron-induced regulation between primary and secondary iron overload. In humans, elevated plasma ferritin was significantly associated with a higher ∆9 desaturase (SCD1) index (C16:1/C16:0) in a cross study of 447 female participants (117). In mice with iron loading, hepatic DG and TG species containing C18:1 represented the majority of the top 20 upregulated glycerolipids, indicating that increased Scd1 expression caused by iron may increase production of monounsaturated fatty acids (118). This was further supported by results from iron-loaded HUVECs, which exhibited increased synthesis of C16:1 and C18:1, compared to saturated counterparts C16:0 and C18:0 demonstrating greater SCD1 activity (37).
The relationship between fatty acid desaturase 1 (FADS1; ∆5 desaturase) and fatty acid desaturase 2 (FADS2; ∆6 desaturase) with iron loading was opposite to that observed for SCD1. Dietary iron loading increased both FADS1 and FADS2 mRNA expression but decreased protein activity (101, 119, 120). These enzymes introduce a double bond at position ∆6 on the acyl chain of linoleic acid (C18:2n-6) and α-linolenic acid (C18:3n-3) (121) and are essential for the synthesis of polyunsaturated fatty acids, such as docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) (122). Taken together, iron’s effects on fatty acid desaturases lead to increased monounsaturated fatty acids and reduced polyunsaturated fatty acids, as observed in plasma and hepatic lipidomic studies (101, 119).
Fatty acid activation
Conversion of fatty acids to their active forms via thioesterification to add CoA to generate fatty acyl-CoA is a required step in fatty acid metabolism (49). For long-chain fatty acids, the reaction is catalysed by the ACS long chain family of enzymes. ACSL1 accounts for ~50% of total hepatic ACSL activity (123) and is known to physically interact with Cpt1 in rat hepatocytes with a potential role in targeting fatty acyl-CoA towards mitochondrial β-oxidation (124). ACSL1 mRNA expression decreased with iron in a dose-dependent manner in HepG2 cells incubated with oleic acid (125) and, in iron-loaded mice, hepatic Acsl1 protein expression was reduced substantially (73). Links between elevated plasma ferritin and reduced ACSL1 gene expression were reported in humans in visceral adipose tissue (126). ACSL3 and ACSL5 gene expression was reduced with iron loading in a dose-dependent manner in primary human hepatocytes (82) and was also decreased in Hjv KO mice (82). These studies indicated a reduction in fatty acid activation and mitochondrial β-oxidation with iron loading. The ACSL4 isozyme preferentially utilises arachidonic acid (C20:4) and overexpression promoted the conversion of arachidonic acid into phosphatidyl ethanolamine (PE), phosphatidyl inositol (PI) and TG (127, 128). Protein expression of Acsl4 was upregulated in the liver of iron-loaded mice (129) and lipidomic investigation of iron-loaded mouse liver indicated increase in PI and TG containing arachidonic acid (118), suggesting increased activity.
Mitochondrial β-oxidation
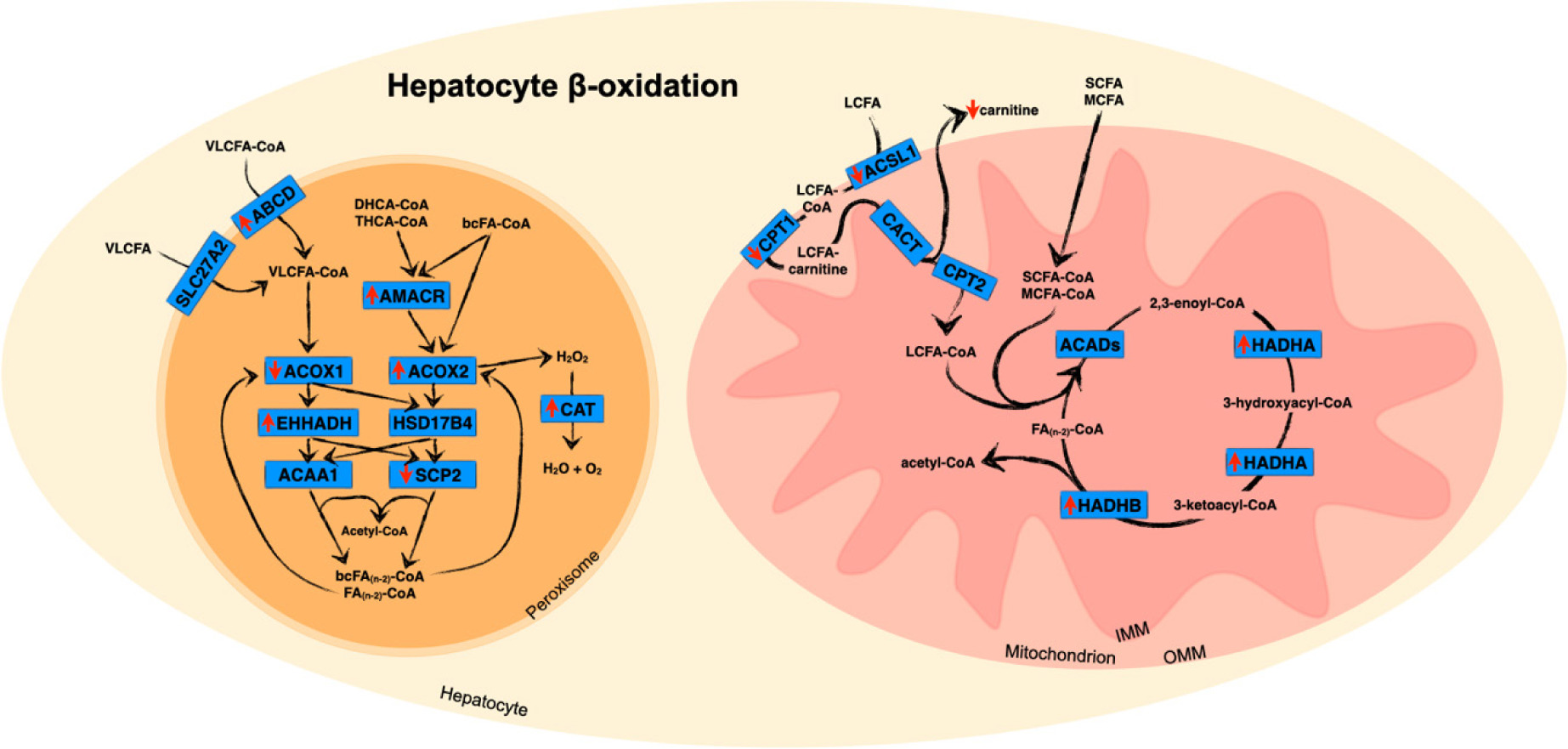
Mitochondrial β-oxidation (Figure 2) is a process that shortens fatty acids into acetyl-CoA, which can be utilised for generating ketone bodies or fully oxidised for energy production via the tricarboxylic acid (TCA) cycle (130, 131). While β-oxidation can also occur in peroxisomes, only the mitochondria are capable of β-oxidation of short- and medium-chain fatty acids (49). Fatty acids must be activated and transported into the mitochondria via the carnitine shuttle (132). As discussed above, iron loading decreases fatty acid activation and significantly downregulates hepatic Cpt1 mRNA and protein expression in genetic and dietary rodent models (35, 36, 53, 83, 105, 133). Mice fed with high fat plus iron also demonstrated decreased Cpt1 mRNA and protein expression, suggesting decreased fatty acid transportation into the mitochondria (45, 105). The activity of rat CPT1 was also reduced with iron loading (101), and L-carnitine and L-acylcarnitine levels decreased in mice following 2 weeks of dietary iron loading (134). Iron loading therefore causes a reduction in mitochondrial fatty acid import, decreasing β-oxidation. While dietary iron loading did not alter CPT2 gene expression (133), genetic iron loading through Hfe KO decreased Cpt2 mRNA (83).
Once within the mitochondria, fatty acids undergo the four steps of β-oxidation (130). The first is dehydrogenation of acyl-CoA to trans-2-enoyl-CoA by one of the acyl-CoA dehydrogenases (ACADs). The last three steps are catalysed by mitochondrial trifunctional enzyme, which comprises two subunits, hydroxyacyl dehydrogenase alpha (HADHA), which completes the 2,3-enoyl-CoA hydratase and the 3-hydroxyacyl-CoA dehydrogenase processes, and hydroxyacyl dehydrogenase beta (HADHB), which completes the 3-ketoacyl-CoA thiolase reaction (130, 135). Iron loading of the human hepatocyte cell line HH4 reduced ACADVL protein, and in iron-loaded mice, Acad10 gene expression was reduced (71), suggesting decreased β-oxidation of very long-chain fatty acids (136). Despite this, protein expression of multiple ACAD isoforms, including Acad10, was not significantly altered in dietary iron-loaded mice. In contrast, HADHA and HADHB proteins were increased, which would be consistent with an increase in mitochondrial β-oxidation (73, 137). The increase in the mitochondrial β-oxidation pathway, coupled with reduced CPT1, may indicate increased utilisation of short-chain fatty acids and decreased use of longer-chain fatty acids for mitochondrial β-oxidation. ApoE KO mice fed with a high fat and high iron diet exhibited reduced Acads and Hadha protein (45), suggesting decreased mitochondrial β-oxidation of short-chain fatty acids and a role for iron in decreasing mitochondrial β-oxidation in NAFLD.
Figure 2: Association between hepatic iron accumulation and dysregulated β-oxidation. Red arrows indicate direction of iron-associated regulation of gene, protein or metabolite expression as discussed in the text. SCFA: short-chain fatty acid; MCFA: medium-chain fatty acid; LCFA: long-chain fatty acid; VLCFA: very long-chain fatty acid; bcAcyl-CoA: branched-chain acyl-CoA; CACT: carnitine acylcarnitine translocase. Other abbreviations are defined in the text. Pathways are based on data from KEGG (181).
Peroxisomal β-oxidation
While the majority of fatty acid β-oxidation is undertaken by the mitochondria, β-oxidation of very long-chain and branched chain fatty acids is dependent on peroxisomes (138). Fatty acids are imported across the peroxisomal membrane via members of the adrenoleukodystrophy (ALD) subfamily of the ATP-binding cassette (ABC) transporters (139, 140). The solute carrier, SLC27A2, is also associated with peroxisomal import (Figure 2) (141, 142). Analogous to mitochondrial β-oxidation, each cycle of peroxisomal β-oxidation requires four enzymatic steps of oxidation, hydration, dehydrogenation and thiolytic cleavage (143). The first reaction in rats and mice is catalysed by one of three isoforms of acyl-CoA oxidase (ACOX) (144). Palmitoyl-CoA oxidase (ACOX1) is specific for saturated and monounsaturated substrates, including very long-chain fatty acids and dicarboxylic fatty acids. ACOX2 specifically reacts with CoA esters of the bile acid intermediates 3α,7α-dihydroxycholestanoic acid (DHCA) and 3α,7α,12α-trihydroxycholestanoic acid (THCA) and branched chain fatty acids, and ACOX3 is active with branched chain fatty acids. The peroxisomal L-bifunctional enzyme (enoyl-CoA hydratase and hydroxyacyl-CoA dehydrogenase [EHHADH] or LBP) and D-bifunctional enzyme (hydroxysteroid-17-beta dehydrogenase [HSD17B4] or DBP) catalyse the second and third steps of peroxisomal β-oxidation. Both have enoyl-CoA hydratase and 3-hydroxyacyl-CoA dehydrogenase activities but with opposite chiral specificity (145). In rodents, the final step is completed by one of three different thiolases—3-ketoacyl-CoA thiolase A or B (Acaa1a or Acaa1b) or sterol carrier protein 2 (Scp2) (143).
There are indications that iron loading perturbs hepatic peroxisomal β-oxidation. Dietary iron-loaded mice exhibited upregulated hepatic gene expression of Abcd2, Ehhadh, and Acot3 (38) as well as protein expression of Acox2, Ehhadh and 2,4-dienoyl-CoA reductase (DECR2), while Scp2 gene and protein expression decreased and Acaa1a and Acaa1b did not change, consistent with an increase in peroxisomal fatty acid uptake and β-oxidation (38, 71, 73, 146). In IONP-injected iron-loaded mice, increased hepatic protein expression of Acox2 and alpha-methylacyl-CoA racemase (Amcr) was reported (104). Catalase (Cat) gene and protein expression was also increased in these mice, suggesting increased generation of hydrogen peroxide, a product of peroxisomal β-oxidation (104). These studies suggest that iron loading is associated with increased hepatic peroxisomal β-oxidation of branched-chain fatty acids and bile acid intermediates. Interestingly, hepatic Acox1 gene expression was decreased in dietary, iron-dextran injected and Hfe KO mice (83, 84), suggesting decreased peroxisomal β-oxidation of straight and monounsaturated fatty acids in both primary and secondary iron loading.
Triglyceride synthesis
Iron overload is frequently associated with increased hepatic TG, leading to elevated hepatocyte lipid droplet formation in both in vitro and in vivo studies (69, 118, 119, 147). Lipid droplets primarily comprise TG and cholesteryl esters, and protect hepatocytes from lipotoxicity (148). Lipidomic investigation of homogenised livers of mice injected with iron dextran demonstrated that iron does not cause the accumulation of only a single type of glycerolipid but instead results in a broader increase in MG, DG, TG and sterol lipids (118). Thus, iron appears to induce lipid accumulation and storage phenotype not only by increasing the synthesis of fatty acids but also by increasing their incorporation into glycerolipids. Glycerol-3-phosphate acyltransferase (GPAT) is the first enzyme of the TG synthesis pathway, producing lysophosphatidic acid (149) (Figure 1). Acylation of lysophosphatidic acid to phosphatidic acid by acylglycerol-3-phosphate O-acyltransferase (AGPAT) is completed in the endoplasmic reticulum (ER) membrane. Subsequent dephosphorylation by phosphohydrate phosphohydrolase (LIPIN) generates DG (149). The final reaction is acylation of DG by diglyceride acyltransferase (DGAT) to form TG. Interestingly, microarray and proteomic studies did not pick up any changes in the expression of genes or proteins associated with the TG synthesis pathway in the presence of iron loading (45). However, activity of GPAT was reduced when incubated with iron sulphate (Fe2+) or iron chloride (Fe3+) (150), raising the question of what caused the TG to accumulate? ApoE KO mice fed with a high fat and high iron diet exhibited no change in Gpat1, Agpat2 or Dgat2 gene and protein expression, potentially due to iron loading decreasing TG synthesis in animals fed with a high fat diet (45). Dietary iron loading similarly reduced hepatic gene expression of Lipin1 in mice (38). HepG2 cells incubated with IONP showed no change in LIPIN1 gene expression; however, iron loading in combination with oleic acid substantially increased LIPIN1 gene expression (47) and this was similarly observed in high fat-fed mice with IONP iron loading. These studies suggest that further investigations are required into the effect of iron on TG synthesis to fully unravel the role of iron.
Cholesterol synthesis
Dysregulated cholesterol metabolism, including increased availability of free cholesterol, is associated with NAFLD progression (151–153). The liver has a fundamental role in de novo cholesterol synthesis, which is highly regulated throughout a multistep process (154) (Figure 1). In brief, synthesis begins with the generation of mevalonate from acetate, itself a multistep process utilising acetyl-CoA acetyltransferase (ACAT) to form acetoacetyl-CoA, which subsequently has an additional acetyl-CoA added by HMG-CoA synthase, generating HMG-CoA (155). The final reaction, catalysed by HMG-CoA reductase (HMGCR), is both rate limiting and the committed step in the process (156, 157). Mevalonate is subsequently converted by several reactions into two activated isoprenes (155). The condensation of multiple activated isoprenes by farnesyl-PP synthase (FDPS) and farnesyl-pyrophosphate farnesyltransferase (FDFT) generates squalene, the precursor of all steroids, a 30-carbon polyunsaturated linear molecule. Squalene undergoes a series of oxidation and reduction reactions to produce the tetracyclic compound, lanosterol, from which cholesterol can be generated via two different, albeit interconnected, routes, termed the Bloch and Kandutsch–Russell pathways (158, 159).
Dietary iron loading was found to upregulate Hmgcr, phosphomevalonate kinase (Pmvk) and Fdps in mice, which may increase production of mevalonate (40, 160). Furthermore, gene expression of Cyp51, delta-14-sterol reductase (Tm7sf2), Hsd17b7, cholestenol delta-isomerase (Ebp) and Sc5d was shown to increase hepatic cholesterol accumulation. Incubation of HUVECs with iron upregulated Hmgcr and Pmvk gene expression and cholesterol biosynthesis (37). Supporting these findings, hydroxymethylglutarate-CoA synthase 2 (Hmgcs2) and Hmgcr protein expression is also increased in dietary iron-loaded mice and rats (73, 88), and hepatic Hmgcr gene expression is upregulated in mice fed with a high fat and high iron diet, demonstrating that iron induced increases in hepatic and plasma cholesterol and cholesteryl esters through increased de novo lipogenesis (118, 161), supporting a role for iron in further elevating cholesterol in NAFLD. While iron loading has been associated with reduced activity of the Hmgcr gene in rats, hepatic cholesterol concentration was not impacted and, as stated by the authors, was consistent with increased oxidative stress, indicative of oxidative damage to the membranes in which the enzymes reside (162).
Approximately 90% of actively metabolised cholesterol is utilised for bile acid synthesis (163). The first reaction is rate-limiting and is catalysed by members of the cytochrome P450 (CYP) family (164). Cyp7a1 gene and protein expression was reduced in the liver of iron-loaded mice and rats (88). It was demonstrated that iron loading reduced the activity of Cyp7a1 in rat hepatic microsomes (162). Iron loading subsequently decreased the synthesis and removal of bile acids by the liver and could be an alternative cause of plasma and hepatic cholesterol accumulation.
Lipid metabolism regulation
Multiple transcription factors are involved in regulating lipid metabolism, of note are sterol regulator element binding proteins (SREBPs) and peroxisome proliferator-activated receptors (PPARs) (165–167). SREBP1c is the primary isoform in the liver and is involved in regulating genes involved in fatty acid synthesis, while SREBP2 activates the cholesterol uptake and synthesis pathway (165). Iron loading was demonstrated to increase the hepatic expression of Srebp1 protein in mice and rats fed with a high iron diet (100, 101, 168). However, rats injected with gleptoferron exhibited no change in gene or hepatic protein expression (88), despite an increase in Srebp2 protein expression. In vitro HUVECs iron loaded with ferric ammonium citrate were reported to elevate SREBP2 gene expression (37). PPARα is highly expressed in the liver and has a fundamental role in upregulating mitochondrial and peroxisomal β-oxidation, ketogenesis and lipoprotein assembly (166–167). Iron loading in rodents, induced either by diet or dextran injection, was shown to reduce hepatic Pparα and Pparγ gene and protein expression (36, 100, 101, 105, 161). These studies support the observed iron-associated alterations to hepatic lipid metabolism, with increased SREBP1 and SREBP2 increasing the expression of lipid biosynthetic genes, and decreased PPARs decreasing the expression of lipid catabolic pathways (35, 100, 101, 133, 169).
Assembly and secretion of very low-density lipoproteins
The final method by which hepatic lipid loading may be decreased is through export. Fatty acid export from the liver is primarily in the form of TG and cholesteryl ester-rich lipoproteins, including very low-density lipoproteins (VLDL) and high-density lipoproteins (HDL), for delivery to muscles for oxidation and to adipose tissue for storage (170, 171). VLDL production is a two-step process beginning in the ER lumen (172) incorporating small quantities of TG on to apoB100 facilitated by microsomal triglyceride transfer protein (MTP) (173). In vitro studies have repeatedly demonstrated iron loading and ferritin post-translationally inhibiting the secretion of ApoB100, leading to increased ER-associated degradation in hepatic cell lines (174–176). Interestingly, while iron dextran-injected mice showed increased Mtp and ApoB100 gene expression (36), rats injected with iron dextran exhibited no change in Mtp gene expression when fed with a control diet or high fat diet (177). Furthermore, dietary iron-loaded mice showed no change in ApoB100 gene expression (40). A recent proteomic study supported no change in hepatic Mtp protein levels in either dietary iron-loaded or Hjv KO mice, suggesting iron did not alter Mtp expression. Similarly, no changes were reported in hepatic ApoB100 with dietary iron-loading in microarray or proteomic studies. Despite this, animal models and humans exhibit an association between iron parameters and plasma VLDL and LDL (69, 178, 179).
Interestingly, multiple studies have reported that iron loading upregulated hepatic expression of other lipoproteins, primarily ApoAIV and ApoCIII (38, 40, 45). Increased hepatic ApoAIV increased TG export by promotion of ApoB100 lipoprotein assembly, and increased VLDL secretion, resulting in raised plasma TG concentrations (180). Similarly, ApoCIII was shown to upregulate VLDL production, suggesting iron loading may increase hepatic lipoprotein production and secretion through increased ApoAIV and ApoCIII expression.
Conclusion
The liver is central to maintaining whole body lipid homeostasis. The role of overnutrition in dysregulating the activity of metabolic pathways leading to common metabolic conditions, including NAFLD, is well understood. It is increasingly clear from the literature that iron loading induces substantial and complex alterations in hepatic lipid metabolism and appears to play a role in both initial development and subsequent progression of NAFLD through increased lipid availability, de novo synthesis, altered lipid composition and dysregulated hepatic lipid catabolism and export. The exact molecular mechanisms by which iron alters hepatic lipid homeostasis are yet to be fully elucidated. Further investigation and understanding of iron, lipids and their interactions are ultimately required to further our understanding of metabolic diseases such as NAFLD.
Acknowledgements
Clinton J Kidman is the recipient of an Australian Government Research Training Programme scholarship. Keea R Inder-Smith is the recipient of an Australian Government Research Training Programme scholarship and an AINSE Ltd Postgraduate Research Award (PGRA). Mark J Hackett is the recipient of an Australian Research Council Future Fellowship.
Author contributions (CRediT)
Clinton J Kidman: conceptualisation, visualisation, writing of original draft, reviewing and editing of manuscript. Cyril DS Mamotte: supervision, writing, reviewing and editing of manuscript. Keea R Inder-Smith: writing, reviewing and editing of manuscript. Mark J Tobin: supervision, reviewing and editing of manuscript. Mark J Hackett: supervision, reviewing and editing of manuscript. Ross M Graham: conceptualisation, supervision, project administration, reviewing and editing of manuscript.
Conflicts of interest
The authors declared no potential conflict of interest with respect to research, authorship and/or publication of this article.
Abbreviations
ABC, ATP binding cassette; ACAA, ketoacyl-CoA thiolase; ACAD, acyl-CoA dehydrogenase; ACC/ACACA, acetyl-CoA carboxylase; ACAT, acetyl-CoA acetyltransferase; ACLY, ATP-citrate lyase; ACOX, acyl-CoA oxidase; ACSL, acyl-CoA synthetase; AGPAT, acylglycerol-3-phosphate-O-acyltransferase; AMACR, alpha-methylacyl-CoA racemase; CAT, catalase; CYP, cytochrome P450; DECR2, dienoyl-CoA reductase; DG, diglyceride; DGAT, diglyceride acyltransferase; DHA, docosahexanoic acid; DNL, de novo lipogenesis; EBP, cholestenol delta-isomerase; EHHADH, enoyl-CoA hydratase and hydroxyacyl-CoA dehydrogenase; ELOVL, elongation of very long chain fatty acids; EPA, eicosapentanoic acid; ER, endoplasmic reticulum; FABP, fatty acid binding proteins; FADS, fatty acid desaturase; FAS, fatty acid synthase; FAT/CD36, fatty acid translocase; FATP, fatty acid transport protein; FDFT, farnesyl-pyrophosphate farnesyltransferase; FDPS, farnesyl-pyrophosphate synthase; FFA, free fatty acids; GPAT, glycerol-3-phosphate acyltransferase; HADHA, hydroxyacyl dehydrogenase alpha; HADHB, hydroxyacyl dehydrogenase beta; HDL, high density lipoprotein; HJV, haemojuvelin; HMGCR, hydroxymethylglutarate-CoA reductase; HMGCS, hydroxymethylglutarate-CoA synthase; HSD17B, hydroxysteroid-17-beta dehydrogenase; HUVEC, human umbilical vein endothelial cells; IONP, iron oxide nanoparticle; KO, knockout; LDL, low density lipoprotein; LDLR, low density lipoprotein receptor; LIPE, hormone sensitive lipase; LIPIN, phosphohydrate phosphohydratase; MG, monoglyceride; MGL, monoglyceride lipase; NAFLD, non-alcoholic fatty liver disease; NTBI, non-transferrin bound iron; PE, phosphoethanolamine; PI, phosphoinositol; PKA, protein kinase A; PMVK, phosphomevalonate kinase; SCD1, stearoyl-CoA desaturase; SCP2, sterol carrier protein 2; SLC27, solute carrier protein 27; TG, triglyceride; TM7SF2, delta-14-sterol reductase; VLDL, very low density lipoprotein.
REFERENCES
1. Srai SK, Sharp P. Proteins of iron homeostasis. In: Anderson GJ, McLaren GD, editors. Iron physiology and pathophysiology in humans. Totowa, NJ: Humana Press; 2012. pp. 3–25. 10.1007/978-1-60327-485-2_1
2. Muckenthaler MU, Lill R. Cellular iron physiology. In: Anderson GJ, McLaren GD, editors. Iron physiology and pathophysiology in humans. Totowa, NJ: Humana Press; 2012. pp. 27–50. 10.1007/978-1-60327-485-2_2
3. Puig S, Ramos-Alonso L, Romero AM, Martínez-Pastor MT. The elemental role of iron in DNA synthesis and repair. Metallomics. 2017;9:1483–500. 10.1039/C7MT00116A
4. Levi S, Rovida E. The role of iron in mitochondrial function. Biochim Biophys Acta. 2009;1790:629–36. 10.1016/j.bbagen.2008.09.008
5. Liochev SI, Fridovich I. Superoxide and iron: Partners in crime. IUBMB Life. 1999;48:157–61. 10.1080/713803492
6. Wallace DF. The regulation of iron absorption and homeostasis. Clin Biochem Rev. 2016;37:51–62.
7. Green R, Charlton R, Seftel H, Bothwell T, Mayet F, Adams B, et al. Body iron excretion in man: A collaborative study. Am J Med. 1968;45:336–53. 10.1016/0002-9343(68)90069-7
8. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–3. 10.1126/science.1104742
9. Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA. 2001;98:8780–85. 10.1073/pnas.151179498
10. Corradini E, Buzzetti E, Pietrangelo A. Genetic iron overload disorders. Mol Aspects Med. 2020;75:100896. 10.1016/j.mam.2020.100896
11. Swanson CA. Iron intake and regulation: Implications for iron deficiency and iron overload. Alcohol 2003;30:99–102. 10.1016/S0741-8329(03)00103-4
12. Zelber-Sagi S, Nitzan-Kaluski D, Halpern Z, Oren R. NAFLD and hyperinsulinemia are major determinants of serum ferritin levels. J Hepatol. 2007;46:700–7. 10.1016/j.jhep.2006.09.018
13. Mendler M-H, Turlin B, Moirand R, Jouanolle A-M, Sapey T, Guyader D, et al. Insulin resistance-associated hepatic iron overload. Gastroenterology. 1999;117:1155–63. 10.1016/S0016-5085(99)70401-4
14. Turlin B, Mendler MH, Moirand R, Guyader D, Guillygomarc’h A, Deugnier Y. Histologic features of the liver in insulin resistance-associated iron overload: A study of 139 patients. Am J Clin Pathol. 2001;116:263–70. 10.1309/WWNE-KW2C-4KTW-PTJ5
15. Nelson JE, Klintworth H, Kowdley KV. Iron metabolism in nonalcoholic fatty liver disease. Curr Gastroenterol Rep. 2012;14:8–16. 10.1007/s11894-011-0234-4
16. Sayiner M, Koenig A, Henry L, Younossi ZM. Epidemiology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in the United States and the rest of the world. Clin Liver Dis. 2016;20:205–14. 10.1016/j.cld.2015.10.001
17. Hudgins LC, Hellerstein M, Seidman C, Neese R, Diakun J, Hirsch J. Human fatty acid synthesis is stimulated by a eucaloric low fat, high carbohydrate diet. J Clin Invest. 1996;97:2081–91. 10.1172/JCI118645
18. Cohen DE, Fisher EA. Lipoprotein metabolism, dyslipidemia, and nonalcoholic fatty liver disease. Sem Liver Dis. 2013;33:380–8. 10.1055/s-0033-1358519
19. Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, et al. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology. 2004;40:1387–95. 10.1002/hep.20466
20. Brunt EM, Tiniakos DG. Histopathology of nonalcoholic fatty liver disease. World J Gastroenterol. 2010;16:5286–96. 10.3748/wjg.v16.i42.5286
21. Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertension Rep. 2018;20:12. 10.1007/s11906-018-0812-z
22. Jaacks LM, Vandevijvere S, Pan A, McGowan CJ, Wallace C, Imamura F, et al. The obesity transition: Stages of the global epidemic. Lancet Diab Endocrinol. 2019;7:231–40. 10.1016/S2213-8587(19)30026-9
23. Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis. 2010;28:155–61. 10.1159/000282080
24. Zhang DL, Ghosh MC, Rouault TA. The physiological functions of iron regulatory proteins in iron homeostasis—An update. Front Pharmacol. 2014;5:124. 10.3389/fphar.2014.00124
25. Van Wyk CP, Linder-Horowitz M, Munro HN. Effect of iron loading on non-heme iron compounds in different liver cell populations. J Biol Chem. 1971;246:1025–31. 10.1016/S0021-9258(18)62426-3
26. Young SP, Roberts S, Bomford A. Intracellular processing of transferrin and iron by isolated rat hepatocytes. Biochem J. 1985;232:819–23. 10.1042/bj2320819
27. Fernández-Real JM, López-Bermejo A, Ricart W. Cross-talk between iron metabolism and diabetes. Diabetes. 2002;51:2348–54. 10.2337/diabetes.51.8.2348
28. Nelson JE, Wilson L, Brunt EM, Yeh MM, Kleiner DE, Unalp-Arida A, et al. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology. 2011;53:448–57. 10.1002/hep.24038
29. Fujita N, Miyachi H, Tanaka H, Takeo M, Nakagawa N, Kobayashi Y, et al. Iron overload is associated with hepatic oxidative damage to DNA in nonalcoholic steatohepatitis. Cancer Epidemiol Biomarkers Prev. 2009;18:424–32. 10.1158/1055-9965.EPI-08-0725
30. Brown KE, Dennery PA, Ridnour LA, Fimmel CJ, Kladney RD, Brunt EM, et al. Effect of iron overload and dietary fat on indices of oxidative stress and hepatic fibrogenesis in rats. Liver Int. 2003;23:232–42. 10.1034/j.1600-0676.2003.00832.x
31. Yin H, Xu L, Porter NA. Free radical lipid peroxidation: Mechanisms and analysis. Chem Rev. 2011;111:5944–72. 10.1021/cr200084z
32. Chevion M. A site-specific mechanism for free radical induced biological damage: The essential role of redox-active transition metals. Free Radic Biol Med. 1988;5:27–37. 10.1016/0891-5849(88)90059-7
33. Violi F, Marino R, Milite MT, Loffredo L. Nitric oxide and its role in lipid peroxidation. Diabetes/Metab Res Rev. 1999;15:283–8. 10.1002/(SICI)1520-7560(199907/08)15:4<283::AID-DMRR42>3.0.CO;2-U
34. Eder SK, Feldman A, Strebinger G, Kemnitz J, Zandanell S, Niederseer D, et al. Mesenchymal iron deposition is associated with adverse long-term outcome in non-alcoholic fatty liver disease. Liver Int. 2020;40:1872–82. 10.1111/liv.14503
35. Handa P, Morgan-Stevenson V, Maliken BD, Nelson JE, Washington S, Westerman M, et al. Iron overload results in hepatic oxidative stress, immune cell activation, and hepatocellular ballooning injury, leading to nonalcoholic steatohepatitis in genetically obese mice. Am J Physiol Gastrointest Liver Physiol. 2016;310:G117–27. 10.1152/ajpgi.00246.2015
36. Silva M, Guerra JFdC, Sampaio AFS, Lima WGd, Silva ME, Pedrosa ML. Iron dextran increases hepatic oxidative stress and alters expression of genes related to lipid metabolism contributing to hyperlipidaemia in murine model. Bio Med Res Int. 2015;2015:272617. 10.1155/2015/272617
37. Fisher AL, Srole DN, Palaskas NJ, Meriwether D, Reddy ST, Ganz T, et al. Iron loading induces cholesterol synthesis and sensitizes endothelial cells to TNFα-mediated apoptosis. J Biol Chem. 2021;297:101156. 10.1016/j.jbc.2021.101156
38. Rodriguez A, Luukkaala T, Fleming RE, Britton RS, Bacon BR, Parkkila S. Global transcriptional response to Hfe deficiency and dietary iron overload in mouse liver and duodenum. PLoS One 2009;4:e7212. 10.1371/journal.pone.0007212
39. Bai S, Luo W, Liu H, Zhang K, Wang J, Ding X, et al. Effects of high dietary iron on the lipid metabolism in the liver and adipose tissue of male broiler chickens. Anim Feed Sci Technol. 2021;282:115131. 10.1016/j.anifeedsci.2021.115131
40. Graham RM, Chua AC, Carter KW, Delima RD, Johnstone D, Herbison CE, et al. Hepatic iron loading in mice increases cholesterol biosynthesis. Hepatology. 2010;52:462–71. 10.1002/hep.23712
41. Kidman CJ, Mamotte CDS, Eynaud MA, Reinhardt J, Vongsvivut J, Tobin MJ, et al. Tracking biochemical changes induced by iron loading in AML12 cells with synchrotron live cell, time-lapse infrared microscopy. Biochem J. 2021;478:1227–39. 10.1042/BCJ20200653
42. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–51. 10.1172/JCI23621
43. Hagenfeldt L, Wahren J, Pernow B, Räf L. Uptake of individual free fatty acids by skeletal muscle and liver in man. J Clin Invest. 1972;51:2324–30. 10.1172/JCI107043
44. Barrows BR, Parks EJ. Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J Clin Endocrinol Metab. 2006;91:1446–52. 10.1210/jc.2005-1709
45. Xiao L, Luo G, Li H, Yao P, Tang Y. Dietary iron overload mitigates atherosclerosis in high-fat diet-fed apolipoprotein E knockout mice: Role of dysregulated hepatic fatty acid metabolism. Biochim Biophys Acta Mol Cell Biol Lipids. 2021;1866:159004. 10.1016/j.bbalip.2021.159004
46. Kim J, Jia X, Buckett PD, Liu S, Lee C-H, Wessling-Resnick M. Iron loading impairs lipoprotein lipase activity and promotes hypertriglyceridemia. FASEB J. 2013;27:1657–63. 10.1096/fj.12-224386
47. Zhu M, Chen H, Zhou S, Zheng L, Li X, Chu R, et al. Iron oxide nanoparticles aggravate hepatic steatosis and liver injury in nonalcoholic fatty liver disease through BMP-SMAD-mediated hepatic iron overload. Nanotoxicology. 2021;15:761–78. 10.1080/17435390.2021.1919329
48. Wlazlo N, van Greevenbroek MMJ, Ferreira I, Jansen EHJM, Feskens EJM, van der Kallen CJH, et al. Iron metabolism is associated with adipocyte insulin resistance and plasma adiponectin: The Cohort on Diabetes and Atherosclerosis Maastricht (CODAM) study. Diab Care. 2013;36:309–15. 10.2337/dc12-0505
49. Alves-Bezerra M, Cohen DE. Triglyceride metabolism in the liver. Compr Physiol. 2017;8:1–8. 10.1002/cphy.c170012
50. Zimmermann R, Strauss Juliane G, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–86. 10.1126/science.1100747
51. Kershaw EE, Hamm JK, Verhagen LAW, Peroni O, Katic M, Flier JS. Adipose triglyceride lipase: Function, regulation by insulin, and comparison with adiponutrin. Diabetes. 2006;55:148–57. 10.2337/diabetes.55.01.06.db05-0982
52. Lafontan M, Langin D. Lipolysis and lipid mobilization in human adipose tissue. Prog Lipid Res. 2009;48:275–97. 10.1016/j.plipres.2009.05.001
53. Katsumura M, Takagi S, Oya H, Tamura S, Saneyasu T, Honda K, et al. Effects of dietary heme iron and exercise training on abdominal fat accumulation and lipid metabolism in high-fat diet-fed mice. Animal Sci J. 2017;88:1100–6. 10.1111/asj.12734
54. Romero AR, Mu A, Ayres JS. Adipose triglyceride lipase mediates lipolysis and lipid mobilization in response to iron-mediated negative energy balance. iScience. 2022;25:103941. 10.1016/j.isci.2022.103941
55. Folgueras AR, Freitas-Rodríguez S, Ramsay AJ, Garabaya C, Rodríguez F, Velasco G, et al. Matriptase-2 deficiency protects from obesity by modulating iron homeostasis. Nature Comm. 2018;9:1350. 10.1038/s41467-018-03853-1
56. Rumberger JM, Peters T, Jr., Burrington C, Green A. Transferrin and iron contribute to the lipolytic effect of serum in isolated adipocytes. Diabetes. 2004;53:2535–41. 10.2337/diabetes.53.10.2535
57. Green A, Basile R, Rumberger JM. Transferrin and iron induce insulin resistance of glucose transport in adipocytes. Metab Clin Exp. 2006;55:1042–5. 10.1016/j.metabol.2006.03.015
58. Ryan BJ, Van Pelt DW, Guth LM, Ludzki AC, Gioscia-Ryan RA, Ahn C, et al. Plasma ferritin concentration is positively associated with in vivo fatty acid mobilization and insulin resistance in obese women. Exp Physiol. 2018;103:1443–7. 10.1113/EP087283
59. Britton LJ, Subramaniam VN, Crawford DH. Iron and non-alcoholic fatty liver disease. World J Gastroenterol. 2016;22:8112–22. 10.3748/wjg.v22.i36.8112
60. Tang Y, Wang D, Zhang H, Zhang Y, Wang J, Qi R, et al. Rapid responses of adipocytes to iron overload increase serum TG level by decreasing adiponectin. J Cell Physiol. 2021;236:7544–53. 10.1002/jcp.30391
61. Qiao L, Kinney B, Schaack J, Shao J. Adiponectin inhibits lipolysis in mouse adipocytes. Diabetes. 2011;60:1519–27. 10.2337/db10-1017
62. Wedellová Z, Dietrich J, Siklová-Vítková M, Kološtová K, Kováčiková M, Dušková M, et al. Adiponectin inhibits spontaneous and catecholamine-induced lipolysis in human adipocytes of non-obese subjects through AMPK-dependent mechanisms. Physiol Res. 2011;60(1):139–48. 10.33549/physiolres.931863
63. Fasshauer M, Klein J, Neumann S, Eszlinger M, Paschke R. Adiponectin gene expression is inhibited by β-adrenergic stimulation via protein kinase A in 3T3-L1 adipocytes. FEBS Lett. 2001;507:142–6. 10.1016/S0014-5793(01)02960-X
64. Simard JR, Kamp F, Hamilton JA. Measuring the adsorption of fatty acids to phospholipid vesicles by multiple fluorescence probes. Biophys J. 2008;94:4493–503. 10.1529/biophysj.107.121186
65. Anderson CM, Stahl A. SLC27 fatty acid transport proteins. Mol Aspects Med. 2013;34:516–28. 10.1016/j.mam.2012.07.010
66. Doege H, Baillie RA, Ortegon AM, Tsang B, Wu Q, Punreddy S, et al. Targeted deletion of FATP5 reveals multiple functions in liver metabolism: Alterations in hepatic lipid homeostasis. Gastroenterology. 2006;130:1245–58. 10.1053/j.gastro.2006.02.006
67. Martius G, Alwahsh SM, Rave-Fränk M, Hess CF, Christiansen H, Ramadori G, et al. Hepatic fat accumulation and regulation of FAT/CD36: An effect of hepatic irradiation. Int J Clin Exp Pathol. 2014;7:5379–92.
68. Falcon A, Doege H, Fluitt A, Tsang B, Watson N, Kay MA, et al. FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase. Am J Physiol Endocrinol Metab. 2010;299:E384–93. 10.1152/ajpendo.00226.2010
69. Mayneris-Perxachs J, Cardellini M, Hoyles L, Latorre J, Davato F, Moreno-Navarrete JM, et al. Iron status influences non-alcoholic fatty liver disease in obesity through the gut microbiome. Microbiome. 2021;9:104. 10.1186/s40168-021-01052-7
70. Wang Hz, Jiang X, Jieyu W, Zhang L, Huang J, Zhang Y, et al. Iron overload coordinately promotes ferritin expression and fat accumulation in Caenorhabditis elegans. Genetics. 2016;203(1):241–53. 10.1534/genetics.116.186742
71. Xiong H, Zhang C, Han L, Xu T, Saeed K, Han J, et al. Suppressed farnesoid X receptor by iron overload in mice and humans potentiates iron-induced hepatotoxicity. Hepatology. 2022;76:387–403. 10.1002/hep.32270
72. Wei Y, Zhao M, Yang F, Mao Y, Xie H, Zhou Q. Iron overload by superparamagnetic iron oxide nanoparticles is a high risk factor in cirrhosis by a systems toxicology assessment. Sci Rep. 2016;6:29110. 10.1038/srep29110
73. Allameh A, Hüttmann N, Charlebois E, Katsarou A, Gu W, Gkouvatsos K, et al. Hemojuvelin deficiency promotes liver mitochondrial dysfunction and predisposes mice to hepatocellular carcinoma. Comm Biol. 2022;5:153. 10.1038/s42003-022-03108-2
74. Smathers RL, Petersen DR. The human fatty acid-binding protein family: Evolutionary divergences and functions. Hum Genom. 2011;5:170. 10.1186/1479-7364-5-3-170
75. Bass NM. The cellular fatty acid binding proteins: Aspects of structure, regulation, and function. Int Rev Cytol. 1988;111:143–84. 10.1016/s0074-7696(08)61733-7
76. McArthur MJ, Atshaves BP, Frolov A, Foxworth WD, Kier AB, Schroeder F. Cellular uptake and intracellular trafficking of long chain fatty acids. J Lipid Res. 1999;40:1371–83. 10.1016/S0022-2275(20)33379-4
77. Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004;40:185–94. 10.1002/hep.20283
78. Sieber J, Jehle A. Free fatty acids and their metabolism affect function and survival of podocytes. Fronti Endocrinol. 2014;5:186. 10.3389/fendo.2014.00186
79. Sarkar-Banerjee S, Chowdhury S, Sanyal D, Mitra T, Roy SS, Chattopadhyay K. The role of intestinal fatty acid binding proteins in protecting cells from fatty acid induced impairment of mitochondrial dynamics and apoptosis. Cell Physiol Biochem. 2018;51:1658–78. 10.1159/000495672
80. Murphy EJ, Prows DR, Stiles T, Schroeder F. Liver and intestinal fatty acid-binding protein expression increases phospholipid content and alters phospholipid fatty acid composition in L-cell fibroblasts. Lipids. 2000;35:729–38. 10.1007/s11745-000-0579-x
81. Wolfrum C, Buhlmann C, Rolf B, Börchers T, Spener F. Variation of liver-type fatty acid binding protein content in the human hepatoma cell line HepG2 by peroxisome proliferators and antisense RNA affects the rate of fatty acid uptake. Biochim Biophys Acta Mol Cell Biol Lipids. 1999;1437:194–201. 10.1016/S1388-1981(99)00008-6
82. Koppe T, Patchen B, Cheng A, Bhasin M, Vulpe C, Schwartz RE, et al. Nicotinamide N-methyltransferase expression decreases in iron overload, exacerbating toxicity in mouse hepatocytes. Hepatol Comm. 2017;1:803–15. 10.1002/hep4.1083
83. Coppin H, Darnaud V, Kautz L, Meynard D, Aubry M, Mosser J, et al. Gene expression profiling of Hfe-/- liver and duodenum in mouse strains with differing susceptibilities to iron loading: Identification of transcriptional regulatory targets of Hfe and potential hemochromatosis modifiers. Genome Biol. 2007;8:R221. 10.1186/gb-2007-8-10-r221
84. Tan TCH, Crawford DHG, Jaskowski LA, Subramaniam VN, Clouston AD, Crane DI, et al. Excess iron modulates endoplasmic reticulum stress-associated pathways in a mouse model of alcohol and high-fat diet-induced liver injury. Lab Invest. 2013;93:1295–312. 10.1038/labinvest.2013.121
85. van de Sluis B, Wijers M, Herz J. News on the molecular regulation and function of hepatic low-density lipoprotein receptor and LDLR-related protein 1. Curr Opin Lipidol. 2017;28:241–7. 10.1097/MOL.0000000000000411
86. Brown MS, Goldstein JL. Lipoprotein receptors in the liver. Control signals for plasma cholesterol traffic. J Clin Invest. 1983;72:743–7. 10.1172/JCI111044
87. Havel RJ. Functional activities of hepatic lipoprotein receptors. Annu Rev Physiol. 1986;48:119–34. 10.1146/annurev.ph.48.030186.001003
88. Prasnicka A, Lastuvkova H, Alaei Faradonbeh F, Cermanova J, Hroch M, Mokry J, et al. Iron overload reduces synthesis and elimination of bile acids in rat liver. Sci Rep. 2019;9:9780. 10.1038/s41598-019-46150-7
89. Mead JR, Irvine SA, Ramji DP. Lipoprotein lipase: Structure, function, regulation, and role in disease. J Mol Med. 2002;80:753–69. 10.1007/s00109-002-0384-9
90. Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: Lipoprotein lipase-and CD36-mediated pathways. J Lipid Res. 2009;50:S86–90. 10.1194/jlr.R800085-JLR200
91. van Bennekum AM, Kako Y, Weinstock PH, Harrison EH, Deckelbaum RJ, Goldberg IJ, et al. Lipoprotein lipase expression level influences tissue clearance of chylomicron retinyl ester. J Lipid Res 1999;40:565–74. 10.1016/S0022-2275(20)32461-5
92. Hellerstein MK, Schwarz JM, Neese RA. Regulation of hepatic de novo lipogenesis in humans. Annu Rev Nutr. 1996;16:523–57. 10.1146/annurev.nu.16.070196.002515
93. Mitsuyoshi H, Yasui K, Harano Y, Endo M, Tsuji K, Minami M, et al. Analysis of hepatic genes involved in the metabolism of fatty acids and iron in nonalcoholic fatty liver disease. Hepatol Res. 2009;39:366–73. 10.1111/j.1872-034X.2008.00464.x
94. Dorn C, Riener M-O, Kirovski G, Saugspier M, Steib K, Weiss TS, et al. Expression of fatty acid synthase in nonalcoholic fatty liver disease. Int J Clin Exp Pathol. 2010;3:505–14.
95. Chakravarty B, Gu Z, Chirala Subrahmanyam S, Wakil Salih J, Quiocho Florante A. Human fatty acid synthase: Structure and substrate selectivity of the thioesterase domain. Proc Natl Acad Sci USA. 2004;101:15567–72. 10.1073/pnas.0406901101
96. Zhao S, Torres A, Henry Ryan A, Trefely S, Wallace M, Lee Joyce V, et al. ATP-citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep. 2016;17:1037–52. 10.1016/j.celrep.2016.09.069
97. Fernandez S, Viola JM, Torres A, Wallace M, Trefely S, Zhao S, et al. Adipocyte ACLY facilitates dietary carbohydrate handling to maintain metabolic homeostasis in females. Cell Rep. 2019;27:2772–84. e2776. 10.1016/j.celrep.2019.04.112
98. Bianchi A, Evans JL, Iverson AJ, Nordlund AC, Watts TD, Witters LA. Identification of an isozymic form of acetyl-CoA carboxylase. J Biol Chem. 1990;265:1502–9. 10.1016/S0021-9258(19)40045-8
99. King MW. Lipids: Chapter 19: Fatty acid synthesis. In: King MW, editor. Integrative medical biochemistry examination and board review. New York, NY: McGraw-Hill Education, 2014. https://accesspharmacy.mhmedical.com/content.aspx?bookid=1696§ionid=111398959
100. Choi JS, Koh IU, Lee HJ, Kim WH, Song J. Effects of excess dietary iron and fat on glucose and lipid metabolism. J Nutr Biochem. 2013;24:1634–44. 10.1016/j.jnutbio.2013.02.004
101. Valenzuela R, Rincón-Cervera, MÁ Echeverría F, Barrera C, Espinosa A, Hernández-Rodas MC, et al. Iron-induced pro-oxidant and pro-lipogenic responses in relation to impaired synthesis and accretion of long-chain polyunsaturated fatty acids in rat hepatic and extrahepatic tissues. Nutrition. 2018;45:49–58. 10.1016/j.nut.2017.07.007
102. Abu-Elheiga L, Matzuk MM, Kordari P, Oh W, Shaikenov T, Gu Z, et al. Mutant mice lacking acetyl-CoA carboxylase 1 are embryonically lethal. Proc Natl Acad Sci USA. 2005;102:12011–16. 10.1073/pnas.0505714102
103. Abu-Elheiga L, Brinkley WR, Zhong L, Chirala SS, Woldegiorgis G, Wakil SJ. The subcellular localization of acetyl-CoA carboxylase 2. Proc Natl Acad Sci USA. 2000;97:1444–9. 10.1073/pnas.97.4.1444
104. Askri D, Cunin V, Ouni S, Béal D, Rachidi W, Sakly M, et al. Effects of iron oxide nanoparticles (γ-Fe2O3) on liver, lung and brain proteomes following sub-acute intranasal exposure: A new toxicological assessment in rat model using iTRAQ-based quantitative proteomics. Int J Mol Sci. 2019;20:5186. 10.3390/ijms20205186
105. Ma W, Jia L, Xiong Q, Du H. Iron overload protects from obesity by ferroptosis. Foods (Basel, Switzerland). 2021;10:1787. 10.3390/foods10081787
106. Guillou H, Zadravec D, Martin PGP, Jacobsson A. The key roles of elongases and desaturases in mammalian fatty acid metabolism: Insights from transgenic mice. Prog Lipid Res. 2010;49:186–99. 10.1016/j.plipres.2009.12.002
107. Gonzalez M, Mutch D. Diet regulation of long-chain PUFA synthesis: Role of macronutrients, micronutrients, and polyphenols on δ-5/δ-6 desaturases and elongases 2/5. Adv Nutr. 2021;12:980–94. 10.1093/advances/nmaa142
108. Tamura K, Makino A, Hullin-Matsuda Fo, Kobayashi T, Furihata M, Chung S, et al. Novel lipogenic enzyme ELOVL7 is involved in prostate cancer growth through saturated long-chain fatty acid metabolism. Cancer Res. 2009;69:8133–40. 10.1158/0008-5472.CAN-09-0775
109. Kitazawa H, Miyamoto Y, Shimamura K, Nagumo A, Tokita S. Development of a high-density assay for long-chain fatty acyl-CoA elongases. Lipids. 2009;44:765–73. 10.1007/s11745-009-3320-8
110. Agbaga M-P, Brush Richard S, Mandal Md Nawajes A, Henry K, Elliott Michael H, Anderson Robert E. Role of Stargardt-3 macular dystrophy protein (ELOVL4) in the biosynthesis of very long chain fatty acids. Proc Natl Acad Sci USA. 2008;105:12843–8. 10.1073/pnas.0802607105
111. Matsuzaka T, Shimano H, Yahagi N, Kato T, Atsumi A, Yamamoto T, et al. Crucial role of a long-chain fatty acid elongase, Elovl6, in obesity-induced insulin resistance. Nat Med. 2007;13:1193–202. 10.1038/nm1662
112. Faradina A, Tseng S-H, Tung T-H, Huang S-Y, Lee Y-C, Skalny AV, et al. High-dose ferric citrate supplementation attenuates omega-3 polyunsaturated fatty acid biosynthesis via downregulating delta 5 and 6 desaturases in rats with high-fat diet-induced obesity. Food Funct. 2021;12:11819–28. 10.1039/D1FO02680A
113. Pigeon C, Legrand P, Leroyer P, Bouriel M, Turlin B, Brissot P, et al Stearoyl coenzyme A desaturase 1 expression and activity are increased in the liver during iron overload. Biochim Biophys Acta Mol Basis Dis. 2001;1535:275–84. 10.1016/S0925-4439(01)00024-2
114. Rodriguez A, Hilvo M, Kytömäki L, Fleming RE, Britton RS, Bacon BR, et al. Effects of iron loading on muscle: Genome-wide mRNA expression profiling in the mouse. BMC Genomics. 2007;8:379. 10.1186/1471-2164-8-379
115. Ntambi James M, Miyazaki M, Stoehr Jonathan P, Lan H, Kendziorski Christina M, Yandell Brian S, et al. Loss of stearoyl–CoA desaturase-1 function protects mice against adiposity. Proc Natl Acad Sci USA. 2002;99:11482–6. 10.1073/pnas.132384699
116. Britton L, Jaskowski L, Bridle K, Santrampurwala N, Reiling J, Musgrave N, et al. Heterozygous Hfe gene deletion leads to impaired glucose homeostasis, but not liver injury in mice fed a high-calorie diet. Physiol Rep. 2016;4:e12837. 10.14814/phy2.12837
117. Wu Y, Baylin A, Colacino JA. Iron, oxidative stress, and δ9 stearoyl-coenzymeA desaturase index (C16:1/C16:0): An analysis applying the national health and nutrition examination survey 2003–04. Curr Dev Nutr. 2017;2:1–8. 10.1093/cdn/nzx001
118. Ding H, Zhang Q, Yu X, Chen L, Wang Z, Feng J. Lipidomics reveals perturbations in the liver lipid profile of iron-overloaded mice. Metallomics. 2021;13:mfab057. 10.1093/mtomcs/mfab057
119. Barrera C, Valenzuela R, Rincón MA, Espinosa A, López-Arana S, González-Mañan D, et al. Iron-induced derangement in hepatic Δ-5 and Δ-6 desaturation capacity and fatty acid profile leading to steatosis: Impact on extrahepatic tissues and prevention by antioxidant-rich extra virgin olive oil. Prostaglandins Leukot Essent Fat Acids. 2020;153:102058. 10.1016/j.plefa.2020.102058
120. Protchenko O, Baratz E, Jadhav S, Li F, Shakoury-Elizeh M, Gavrilova O, et al. Iron chaperone poly rC binding protein 1 protects mouse liver from lipid peroxidation and steatosis. Hepatology 2021;73:1176–93. 10.1002/hep.31328
121. de Antueno RJ, Knickle LC, Smith H, Elliot ML, Allen SJ, Nwaka S, et al. Activity of human Δ5 and Δ6 desaturases on multiple n-3 and n-6 polyunsaturated fatty acids. FEBS Lett. 2001;509:77–80. 10.1016/S0014-5793(01)03135-0
122. Nakamura MT, Nara TY. Structure, function, and dietary regulation of δ6, δ5, and δ9 desaturases. Annu Rev Nutr. 2004;24:345–76. 10.1146/annurev.nutr.24.121803.063211
123. Li LO, Ellis JM, Paich HA, Wang S, Gong N, Altshuller G, et al. Liver-specific loss of long chain acyl-CoA synthetase-1 decreases triacylglycerol synthesis and beta-oxidation and alters phospholipid fatty acid composition. J Biol Chem. 2009;284:27816–26. 10.1074/jbc.M109.022467
124. Lee K, Kerner J, Hoppel CL. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J Biol Chem. 2011;286:25655–62. 10.1074/jbc.M111.228692
125. Wu Y, Ye Q, Zheng Q, Zhang L, Zhao Y. Study of synergistic effect of free fatty acid and iron on the establishment of nonalcoholic fatty liver disease model. Zhonghua Yu Fang Yi Xue Za Zhi. 2014;48:904–8.
126. Segrestin B, Moreno-Navarrete JM, Seyssel K, Alligier M, Meugnier E, Nazare J-A, et al. Adipose tissue expansion by overfeeding healthy men alters iron gene expression. J Clin Endocrinol Metab. 2019;104:688–96. 10.1210/jc.2018-01169
127. Golej DL, Askari B, Kramer F, Barnhart S, Vivekanandan-Giri A, Pennathur S, et al. Long-chain acyl-CoA synthetase 4 modulates prostaglandin E2 release from human arterial smooth muscle cells. J Lipid Res. 2011;52:782–93. 10.1194/jlr.M013292
128. Cooke M, Orlando U, Maloberti P, Podestá EJ, Cornejo Maciel F. Tyrosine phosphatase SHP2 regulates the expression of acyl-CoA synthetase ACSL4. J Lipid Res. 2011;52:1936–48. 10.1194/jlr.M015552
129. Wu S, Yang J, Sun G, Hu J, Zhang Q, Cai J, et al. Macrophage extracellular traps aggravate iron overload-related liver ischaemia/reperfusion injury. Br J Pharmacol. 2021;178:3783–96. 10.1111/bph.15518
130. Wanders RJA, Ruiter JPN, Ijlst L, Waterham HR, Houten SM. The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J Inherit Metab Dis. 2010;33:479–94. 10.1007/s10545-010-9104-8
131. Adeva-Andany MM, Carneiro-Freire N, Seco-Filgueira M, Fernández-Fernández C, Mouriño-Bayolo D. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion. 2019;46:73–90. 10.1016/j.mito.2018.02.009
132. McGarry JD, Brown NF. The mitochondrial carnitine palmitoyltransferase system—From concept to molecular analysis. Eur J Biochem. 1997;244:1–14. 10.1111/j.1432-1033.1997.00001.x
133. Nishina S, Korenaga M, Hidaka I, Shinozaki A, Sakai A, Gondo T, et al. Hepatitis C virus protein and iron overload induce hepatic steatosis through the unfolded protein response in mice. Liver Int. 2010;30:683–92. 10.1111/j.1478-3231.2010.02210.x
134. Volani C, Paglia G, Smarason SV, Pramstaller PP, Demetz E, Pfeifhofer-Obermair C, et al. Metabolic signature of dietary iron overload in a mouse model. Cells. 2018;7:264. 10.3390/cells7120264
135. Nassir F, Ibdah JA. Role of mitochondria in nonalcoholic fatty liver disease. Int J Mol Sci. 2014;15:8713–42. 10.3390/ijms15058713
136. Li X, Li S, Lu M, Yang G, Shen Y, Zhou X. Proteomic profiling of iron overload-induced human hepatic cells reveals activation of TLR2-mediated inflammatory response. Molecules (Basel, Switzerland). 2016;21:322. 10.3390/molecules21030322
137. Yamamoto K, Abe S, Honda A, Hashimoto J, Aizawa Y, Ishibashi S, et al. Fatty acid beta oxidation enzyme HADHA is a novel potential therapeutic target in malignant lymphoma. LabInvest. 2020;100:353–62. 10.1038/s41374-019-0318-6
138. Waterham HR, Ferdinandusse S, Wanders RJA. Human disorders of peroxisome metabolism and biogenesis. Biochim Biophys Acta Mol Cell Res. 2016;1863:922–33. 10.1016/j.bbamcr.2015.11.015
139. Wanders RJA, Visser WF, van Roermund CWT, Kemp S, Waterham HR. The peroxisomal ABC transporter family. Pflügers Archiv Eur J Physiol. 2007;453:719–34. 10.1007/s00424-006-0142-x
140. van Roermund CWT, Visser WF, Ijlst L, Waterham HR, Wanders RJA. Differential substrate specificities of human ABCD1 and ABCD2 in peroxisomal fatty acid β-oxidation. Biochim Biophys Acta Mol Cell Biol Lipids. 2011;1811:148–52. 10.1016/j.bbalip.2010.11.010
141. Steinberg SJ, Wang SJ, Kim DG, Mihalik SJ, Watkins PA. Human very-long-chain acyl-CoA synthetase: Cloning, topography, and relevance to branched-chain fatty acid metabolism. Biochem Biophys Res Comm. 1999;257:615–21. 10.1006/bbrc.1999.0510
142. Wanders RJA, Waterham HR, Ferdinandusse S. Metabolic interplay between peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Front Cell Dev Biol. 2016;3:83. 10.3389/fcell.2015.00083
143. Poirier Y, Antonenkov VD, Glumoff T, Hiltunen JK. Peroxisomal β-oxidation—A metabolic pathway with multiple functions. Biochim Biophys Acta Mol Cell Res. 2006;1763:1413–26. 10.1016/j.bbamcr.2006.08.034
144. Wanders RJA, Denis SW, Dacremont G. Studies on the substrate specificity of the inducible and non-inducible acyl-CoA oxidases from rat kidney peroxisomes. J Biochem. 1993;113:577–82. 10.1093/oxfordjournals.jbchem.a124086
145. Qin Y-M, Haapalainen AM, Kilpeläinen SH, Marttila MS, Koski MK, Glumoff T, et al. Human peroxisomal multifunctional enzyme type 2: Site-directed mutagenesis studies show the importance of two protic residues for 2-enoyl-coA hydratase 2 activity. J Biol Chem. 2000;275:4965–72. 10.1074/jbc.275.7.4965
146. Moon MS, McDevitt EI, Zhu J, Stanley B, Krzeminski J, Amin S, et al. Elevated hepatic iron activates nf-e2–related factor 2–regulated pathway in a dietary iron overload mouse model. Toxicol Sci. 2012;129:74–85. 10.1093/toxsci/kfs193
147. Kim S-H, Yadav D, Kim S-J, Kim J-R, Cho K-H. High consumption of iron exacerbates hyperlipidemia, atherosclerosis, and female sterility in zebrafish via acceleration of glycation and degradation of serum lipoproteins. Nutrients. 2017;9:690. 10.3390/nu9070690
148. Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019;20:137–55. 10.1038/s41580-018-0085-z
149. Takeuchi K, Reue K. Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am J Physiol Endocrinol Metab. 2009;296:E1195–209. 10.1152/ajpendo.90958.2008
150. Thomas PD, Poznansky MJ. Lipid peroxidation inactivates rat liver microsomal glycerol-3-phosphate acyl transferase. Effect of iron and copper salts and carbon tetrachloride. J Biol Chem. 1990;265:2684–91. 10.1016/S0021-9258(19)39856-4
151. Yasutake K, Nakamuta M, Shima Y, Ohyama A, Masuda K, Haruta N, et al. Nutritional investigation of non-obese patients with non-alcoholic fatty liver disease: The significance of dietary cholesterol. Scand J Gastroenterol. 2009;44:471–7. 10.1080/00365520802588133
152. Musso G, Gambino R, De Michieli F, Cassader M, Rizzetto M, Durazzo M, et al. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology. 2003;37:909–16. 10.1053/jhep.2003.50132
153. Musso G, Gambino R, Cassader M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog Lipid Res. 2013;52:175–91. 10.1016/j.plipres.2012.11.002
154. Trapani L Segatto M Pallottini V. Regulation and deregulation of cholesterol homeostasis: The liver as a metabolic “power station”. World J Hepatol. 2012;4:184–90. 10.4254/wjh.v4.i6.184
155. Cerqueira NMFSA, Oliveira EF, Gesto DS, Santos-Martins D, Moreira C, Moorthy HN, et al. Cholesterol biosynthesis: A mechanistic overview. Biochemistry. 2016;55:5483–506. 10.1021/acs.biochem.6b00342
156. DeBose-Boyd RA. Feedback regulation of cholesterol synthesis: Sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 2008;18:609–21. 10.1038/cr.2008.61
157. Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–30. 10.1038/343425a0
158. Bloch K. The biological synthesis of cholesterol. Science. 1965;150:19–28. 10.1126/science.150.3692.19
159. Kandutsch AA, Russell AE. Preputial gland tumor sterols: III. A metabolic pathway from lanosterol to cholesterol. J Biol Chem. 1960;235:2256–61. 10.1016/S0021-9258(18)64608-3
160. Boyer JL. Bile formation and secretion. Compr Physiol. 2013;3:1035–78. 10.1002/cphy.c120027
161. Padda RS, Gkouvatsos K, Guido M, Mui J, Vali H, Pantopoulos K. A high-fat diet modulates iron metabolism but does not promote liver fibrosis in hemochromatotic Hjv−/− mice. Am J Physiol Gastrointest Liver Physiol. 2014;308:G251–61. 10.1152/ajpgi.00137.2014
162. Brunet S, Thibault L, Delvin E, Yotov W, Bendayan M, Levy E. Dietary iron overload and induced lipid peroxidation are associated with impaired plasma lipid transport and hepatic sterol metabolism in rats. Hepatology. 1999;29:1809–17. 10.1002/hep.510290612
163. Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–74. 10.1146/annurev.biochem.72.121801.161712
164. Chiang JYL. Bile acids: Regulation of synthesis. J Lipid Res. 2009;50:1955–66. 10.1194/jlr.R900010-JLR200
165. DeBose-Boyd RA, Ye J. SREBPs in lipid metabolism, insulin signaling, and beyond. Trends Biochem Sci. 2018 May;43(5):358–68. 10.1016/j.tibs.2018.01.005
166. McMullen PD, Bhattacharya S, Woods CG, Sun B, Yarborough K, Ross SM, et al. A map of the PPARα transcription regulatory network for primary human hepatocytes. Chem Biol Interact. 2014 Feb 25;209:14–24. 10.1016/j.cbi.2013.11.006
167. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. 2015 Mar;62(3):720–33. 10.1016/j.jhep.2014.10.039
168. Dongiovanni P, Ruscica M, Rametta R, Recalcati S, Steffani L, Gatti S, et al. Dietary iron overload induces visceral adipose tissue insulin resistance. Am J Pathol. 2013;182:2254–63. 10.1016/j.ajpath.2013.02.019
169. Petrak J, Myslivcova D, Man P, Cmejla R, Cmejlova J, Vyoral D, et al. Proteomic analysis of hepatic iron overload in mice suggests dysregulation of urea cycle, impairment of fatty acid oxidation, and changes in the methylation cycle. Am J Physiol Gastrointest Liver Physiol. 2007 Jun;292(6):G1490–8. 10.1152/ajpgi.00455.2006
170. Gibbons GF, Wiggins D, Brown AM, Hebbachi AM. Synthesis and function of hepatic very-low-density lipoprotein. Biocheml Soc Trans. 2004;32:59–64. 10.1042/bst0320059
171. Zhou L, Li C, Gao L, Wang A. High-density lipoprotein synthesis and metabolism. Mol Med Rep 2015;12:4015–21. 10.3892/mmr.2015.3930
172. Alexander CA, Hamilton RL, Havel RJ. Subcellular localization of B apoprotein of plasma lipoproteins in rat liver. J Cell Biol. 1976;69:241–63. 10.1083/jcb.69.2.241
173. Wetterau JR, Lin MCM, Jamil H. Microsomal triglyceride transfer protein. Biochimica et Biophysica Acta (BBA) Lipids Lipid Metab. 1997;1345:136–50. 10.1016/S0005-2760(96)00168-3
174. Hevi S, Chuck SL. Ferritins can regulate the secretion of apolipoprotein B. J Biol Chem. 2003;278:31924–29. 10.1074/jbc.M303081200
175. Mancone C, Montaldo C, Santangelo L, Di Giacomo C, Costa V, Amicone L, et al. Ferritin heavy chain is the host factor responsible for HCV-induced inhibition of apoB-100 production and is required for efficient viral infection. J Proteome Res. 2012;11:2786–97. 10.1021/pr201128s
176. Barisani D, Meneveri R, Ginelli E, Cassani C, Conte D. Iron overload and gene expression in HepG2 cells: Analysis by differential display. FEBS Lett. 2000;469:208–12. 10.1016/S0014-5793(00)01280-1
177. Ahmed U, Redgrave T, Oates P. Body iron stores increase hepatic and serum lipid in rats fed a standard western diet. GSTF J Adv Med Res. 2014;1:2. 10.5176/2345-7201_1.2.18
178. Martínez-Soto JM, Candia-Plata MdC, López-Soto LF, Soto-Guzmán JA, Camacho-Villa AY, Álvarez-Hernández G, et al. Increased serum ferritin is associated with oxidized low-density lipoprotein in pre-diabetes patients: A pilot study. Heliyon. 2021;7:e06720. 10.1016/j.heliyon.2021.e06720
179. Wu W, Yuan J, Shen Y, Yu Y, Chen X, Zhang L, et al. Iron overload is related to elevated blood glucose levels in obese children and aggravates high glucose-induced endothelial cell dysfunction in vitro. BMJ Open Diab Res Care. 2020;8:e001426. 10.1136/bmjdrc-2020-001426
180. VerHague MA, Cheng D, Weinberg RB, Shelness GS. Apolipoprotein A-IV expression in mouse liver enhances triglyceride secretion and reduces hepatic lipid content by promoting very low density lipoprotein particle expansion. Arterioscler Thromb Vasc Biol. 2013;33:2501–8. 10.1161/atvb.33.suppl_1.A4
181. Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023 Jan 6;51(D1):D587–92. 10.1093/nar/gkac963
182. Mazein A, Watterson S, Hsieh WY, Griffiths WJ, Ghazal P. A comprehensive machine-readable view of the mammalian cholesterol biosynthesis pathway. Biochem Pharmacol. 2013 Jul 1;86(1):56–66. 10.1016/j.bcp.2013.03.021