
Download
Original Article: Hepatology
Acute Alcohol Tissue Damage: Protective Properties of Betaine
Lucy Petagine1, Hannah Everitt1, Victor R. Preedy2, Roy A. Sherwood3, Vinood B. Patel1, 2*
1Centre for Nutraceuticals, School of Life Sciences, University of Westminster, London, UK;
2Nutrition and Dietetics, King’s College London, London, UK;
3Department of Clinical Biochemistry, King’s College Hospital, London, UK
Abstract
Teenage binge drinking is a major health issue; however, there is a paucity of data on liver injury. Herein, we investigated how acute ethanol affects juvenile hepatic cells through changes in oxidative stress, apoptosis, and liver function, as well as the ability of betaine, which can replenish the antioxidant glutathione and mitigate oxidative injury. Juvenile male Wistar rats were given either water or betaine (2% w/v) for 6 days and treated with either saline 0.15 mol/L NaCl or ethanol (75 mmol/kg bodyweight). After 24 h, liver enzymes, oxidative damage, apoptosis, and parameters of antioxidant enzyme activity were examined. Acute ethanol increased hepatic enzymes (99%, P < 0.05). Total protein and albumin levels were reduced by 14 and 18% (P < 0.001), respectively, which was prevented by betaine treatment. Cytosolic cytochrome c increased by 59% (P < 0.05), corresponding to a decrease in mitochondrial cytochrome c content, which was ameliorated with betaine. Cytosolic glutathione peroxidase was reduced with alcohol (P < 0.05) and was prevented with betaine. Subtle changes were observed in catalase, superoxide dismutase, glutathione reductase, and complex I activity after ethanol treatment. In summary, whilst juvenile animals appear to have higher basal levels of antioxidant enzymes, betaine conferred some protection against alcohol-induced oxidative stress.
Key words: adolescents, antioxidants, binge drinking, liver, oxidative stress
Received: 2 February 2021; Accepted after Revision: 3 March 2021; Published: 19 March 2021.
Author for correspondence: Dr. Vinood Patel, School of Life Sciences, University of Westminster, 115 New Cavendish Street, London W1W 6UW, UK. Email: v.b.patel@westminster.ac.uk
How to cite: Petagine L, et al. Acute Alcohol Tissue Damage: Protective Properties of Betaine. J Ren Hepat Disord. 2021;5(1): 19–29.
Doi: http://dx.doi.org/10.15586/jrenhep.v5i1.96
Copyright: Petagine L, et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Worldwide, alcoholic liver disease (ALD) is a major cause of chronic liver disease, which develops due to excessive, prolonged consumption of alcohol. This results in the formation of fatty liver, which can develop to steatohepatitis, cirrhosis/fibrosis, and lastly hepatocellular cancer (1).
ALD is traditionally associated with the adult population; however, recent statistical reports have identified an alarming increase in the percentage of teenage binge drinking. The proportion of adolescents who binge drink remains high with a shift toward young people drinking more frequently with the intention of becoming intoxicated (2). Adolescents are now exposing their livers to the damaging effects of ethanol more frequently and more seriously than at any time in the past. The United Kingdom, in particular, is consistently categorized as a high prevalence country for underage drinking (3), with a third of British teenagers reporting intoxication by the age of 13 (4). The National Survey on Drug Use and Health has estimated that in the United States 22% of adolescents used alcohol in 2016 (5), whereas the European School Survey Project on Alcohol and Other Drugs has revealed that at least half of the students in three-quarters of the countries surveyed had drunk an alcoholic beverage at age 13 or younger (3, 4). Whilst the clinical consequences of ethanol intake have been studied in adults, it remains unclear as to whether adolescents are more susceptible to ethanol-mediated liver damage, or if they can withstand the adverse effects better than adults. Animal studies with binge ethanol treatment have shown to decrease adenosine triphosphate (ATP) production as well as increase reactive oxygen species (ROS) and mitochondrial dysfunction, thus altering the electron transport chain components, resulting in mitochondrial failure (6–8).
Antioxidant enzymes are capable of catalyzing the decomposition of ROS (9). The three main antioxidant enzymes are superoxide dismutase (SOD), which can protect against toxic effects of superoxide radicals (10); glutathione peroxidase (GPx), which protects against oxidative injury (11); and catalase, which promotes the breakdown of hydrogen peroxide H2O2 (11).
The use of antioxidants serves as a prospective therapeutic approach for the treatment of ALD. Glutathione (GSH) is an intracellular antioxidant, which can neutralize basal levels of ROS (12). Other nutrients such as betaine play a role in the cysteine production pathway (13), ultimately forming GSH. Recent chronic experimental studies have examined the protective effects of betaine against alcohol-induced liver damage (14–18). In animal studies, dietary betaine has been shown to ameliorate the adverse effects of acute (19) and chronic ethanol dosing on liver steatosis and oxidative stress (20–23).
However, there are few studies exploring the effect of ethanol intoxication in the teenage population. Therefore, we aimed to investigate how ethanol affects hepatic cells through changes in oxidative damage, apoptosis, and liver function in juvenile rats. The effects of betaine supplementation on ethanol-induced injury in very young animal models is also limited. The use of betaine will determine if any such damage (and by extension, protection) is mediated through oxidative damage and the antioxidant capacity of the liver.
Materials and Methods
Animals
Juvenile male Wistar rats (50–55 g) were obtained from Charles Rivers (Bicester, UK) and housed according to good laboratory practice guidelines at the Biological Services Unit at Kings College, London. Animals were age and weight matched and divided into four groups: (1) Control, i.p. treated 0.15 mol/L NaCl; (2) Ethanol, i.p. treated ethanol (75 mmol/kg bodyweight); (3) Betaine 2% (w/v) in drinking water and i.p. treated 0.15 mol/L NaCl; and (4) Betaine (2% w/v) in drinking water and i.p. treated ethanol (75 mmol/kg bodyweight). Groups 1 and 2 were given water for 1 week and groups 3 and 4 received free access to food and freshly prepared betaine (2% w/v) in the drinking water. On the 7th day, animals were i.p. treated with either saline or ethanol. Following treatment, food was removed, and the rats were sacrificed 24 h later. Hepatic cytosol and mitochondria were prepared as previously described (24). Blood serum was collected and stored for subsequent liver function test analysis. Liver function tests were measured by standard laboratory diagnostic procedures as previously described (25).
Glutathione levels
GSH levels were determined using an assay adapted from Tietze (26) based on the conversion of 5’5’-dithio-bis-2-(nitrobenzoic acid) (DNTB) to 5-thio-2-nitrobenzoic acid (TNB) by glutathione reductase (GR). Cytosolic or mitochondrial protein (5 µL) was added to the reaction buffer (100 mM sodium phosphate, 1 mM EDTA, 0.5 mM DTNB, 0.175 mM NADPH, 1.7U/mL GR) and the absorbance measured at 412 nm for 25 min.
Malondialdehyde levels
Malondialdehyde (MDA) levels were detected using a colorimetric thiobarbituric acid reactive substances assay adapted from Bar-Or et al (27). Protein (500 µg) was mixed with 400 µL of 20 mM phosphate buffer (pH 7.4) and 500 µL reaction buffer (5 mg/mL thiobarbituric acid, 25 mM NaOH, 50% glacial acetic acid). Samples were tightly sealed and boiled for 1 h, rested on ice for 10 min, and the absorbance measured at 532 nM.
Cytochrome c levels
The expression of Cytochrome c protein was quantified by an immunoblotting technique. Protein (60 µg cytosol or 40 µg mitochondria) was loaded onto gels and following transfer, the membranes were incubated with mouse anti-cytochrome c (1:1000) overnight, followed by the secondary antibody rabbit anti-mouse (1:10,000). Signals were detected using Pierce ECL reagent (Thermofisher, UK). The resultant images were analyzed using the Biorad GS-800 Calibrated Densitometer.
Caspase-3 activity
Caspase-3 activity was evaluated by measuring the fluorescence of N-Acetyl-Asp-Glu-Val-Asp-7-amido-4-trifluoromethylcoumarin (Ac-DEVD-AFC) after cleavage by caspase-3 to 7-amino-4-trifluoromethyl-coumarin (AFC) following incubation with 25 µg of protein. Samples (10 μL) were incubated in darkness with 100 μL of the substrate solution (10 µg/mL Ac-DEVD-AFC, 100 mM HEPES, 10 mM DTT) for 1 h at 37°C. The fluorescence was measured using an excitation wavelength of 380 nm and emission wavelength of 520 nm (28).
Complex I activity
The Complex I Enzyme Activity Microplate Assay Kit (Abcam, UK) was used to determine the activity of mitochondrial OXPHOS Complex I in the electron transport chain. Assay solution was prepared with 40 mM NADH. This method determined whether treatment of cells causes damage at complex I. Absorbance was read at OD 450 nm.
Antioxidant enzyme activity
The Glutathione Peroxidase Cellular Activity Assay Kit (Sigma, UK) was used as an indirect determination method based on the oxidation of GSH to GSSG. The decrease in NADPH absorbance was measured at 340 nm, indicating GPx activity. Glutathione Reductase Assay Kit (Sigma, UK) was used to measure the activity of GR, analyzed by spectrophotometric measurement. The activity was measured by any increase in absorbance caused by the reduction of DTNB at 412 nm. The Catalase Assay Kit (Abcam, UK) was used to measure catalase activity. Unconverted H2O2 reacts with OxiRedTM probe, which was analyzed by spectrophotometric measurement at 570 nm. The SOD Assay Kit-WST (Sigma, UK) was used to measure the activity of SOD via the utilization of Dojindo’s highly water-soluble tetrazolium salt, WST-1 (2-(4-Iodophenyl)- 3-(4-nitrophenyl)-5-(2,4-disulfophenyl)- 2H-tetrazolium,monosodium salt) that produces a water-soluble formazan dye upon reduction with a superoxide anion. SOD activity was measured spectrophotometrically at 440 nm.
Statistical analysis
Results were analyzed using a one-way ANOVA. Data are presented as mean + SEM (n = 3–8) and P < 0.05 was considered statistically significant.
Results
Food, water, and body parameters
For the period of betaine treatment, food, water consumption, and body weights were recorded (Table 1). Betaine had no effect on food or water intake in the 6 days preceding the ethanol treatment, and body weights were also unchanged on the final day. On the other hand, liver weights were increased by 13 and 17% (P < 0.05), respectively, in the ethanol and betaine-/ethanol-treated groups, when compared to their corresponding controls. The ratio of liver weight to body weight was used to account for any differences in individual rat weights and liver sizes. A slight increase was observed in the liver weight/body weight ratio in ethanol-treated animals when compared to their corresponding control. In the ethanol group, a slight increase of 6% was observed, while there was a 5% increase in the betaine plus ethanol group, with the latter significantly increasing by 12% when compared to control animals (P < 0.01) (Table 1).
Table 1: The effect of betaine on food and water intake, body weights, and liver weights.
| Control | Ethanol | Betaine | Betaine + Ethanol | |
|---|---|---|---|---|
| Food intake (g/ rat/ day) | 12.2 ± 0.4 | 11.8 ± 0.6 | 11.0 ± 0.4 | 10.7 ± 0.4 |
| Water intake preinjections (mL/rat/day) | 21.7 ± 1.1 | 21.4 ± 1.2 | 24.1 ± 1.6 | 21.9 ± 1.6 |
| Water intake (post-injections (mL/rat/day) | 20.5 ± 1.4 | 20.0 ± 1.6 | 16.5 ± 1.9* | 17.5 ± 1.3* |
| Final body weight (g) | 72.8 ± 1.6 | 77.3 ± 2.4 | 69.8 ± 1.6 | 75.6 ± 2.1 |
| Liver weight (g) | 2.67 ± 0.0 | 3.01 ± 0.1 | 2.69 ± 0.1 | 3.15 ± 0.2* |
| Liver weight (g)/body weight (kg) | 36.6 ± 0.3 | 39.0 ± 0.8 | 38.4 ± 1.0 | 41.5 ± 1.0** |
During the pretreatment stage, animals were housed in groups in cages and both food and water intake was monitored daily. During the treatment, animals were caged singly, deprived of food but allowed free access to water (either control or supplemented with 2% betaine). The body weights were recorded on the morning of sacrifice and the liver weights were recorded as soon as they were excised. *P < 0.05, **P < 0.01 compared to overall control.
Liver function
Following ethanol treatment, ALT levels increased by 99% (P < 0.05) and AST levels showed an increase of 10% (Table 2). Although betaine alone showed no effect, betaine followed by ethanol led to an increase of 160% (P < 0.001) in ALT levels and 82% (P < 0.05) in AST levels. Following ethanol treatment, the AST/ALT value decreased by 41% (P < 0.001), and decreased by 32% (P < 0.001) following betaine and ethanol treatment. Total protein and albumin levels were also reduced following ethanol treatment by 14% (P < 0.001) and 18% (P < 0.001), respectively. However, this decrease was completely prevented in the betaine and ethanol group when compared to both control and betaine-alone treatment groups. There were no changes in circulating globulin levels in all treatment groups (Table 2).
Table 2: The effect of ethanol and betaine on liver function.
| Treatment | ALT (U/L) | AST (U/L) | AST/ALT | Total Protein (g/L) | Albumin (g/L) | Globulin (g/L) |
|---|---|---|---|---|---|---|
| Control | 59.7 ± 5.6 | 382 ± 43 | 6.4 ± 0.5 | 54.0 ± 1.3 | 36.3 ± 0.8 | 17.7 ± 0.6 |
| Ethanol | 119 ± 15* | 423 ± 34 | 3.8 ± 0.2*** | 46.5 ± 1.4** | 29.6 ± 0.9*** | 16.9 ± 0.7 |
| Betaine | 51.8 ± 2.3 | 313 ± 26 | 6.0 ± 0.3 | 52.3 ± 0.9 | 35.0 ± 0.7 | 17.3 ± 0.3 |
| Betaine/Ethanol | 135 ± 4.2*** | 569 ± 37* | 4.1 ± 0.2*** | 52.3 ± 1.8 | 33.7 ± 0.9 | 18.6 ± 0.9 |
The betaine-supplemented experimental animals were given drinking water containing 2% betaine (w/v) for 6 days. Saline (0.15 mol/L NaCl) or ethanol (75 mmol/ kg bodyweight) was administered i.p. on day 7, and 24 h later the animals were sacrificed. Serum was analyzed by standard biochemistry tests. ALT = alanine aminotransferase; AST = aspartate aminotransferase. Values are mean ± SEM (n = 5–8). *P < 0.05; **P < 0.01; ***P < 0.001 compared to relevant control values.
Apoptosis
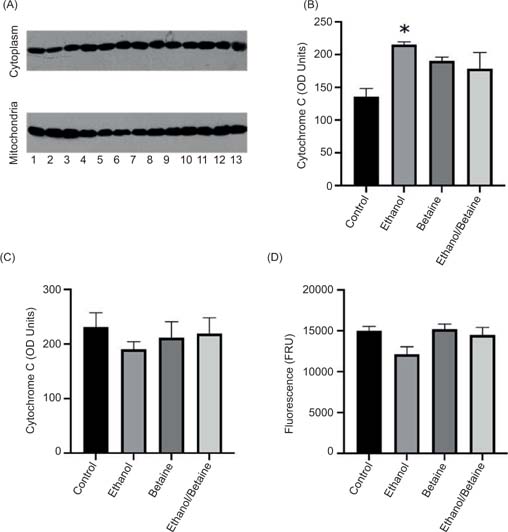
Ethanol exposure led to a significant increase (59%; P < 0.05) in cytosolic cytochrome c levels in comparison to controls. However, cytochrome c release was prevented by betaine pretreatment followed by ethanol (6% decrease) (Figure 1B–C). In the mitochondria, a contrasting pattern occurred following ethanol treatment, whereby cytochrome c levels were lower (18%), confirming the release into the cytosol. Similarly, there was no change in the betaine-alone or betaine-ethanol exposed groups (Figure 1).
Figure 1: The effect of betaine on ethanol-induced changes in (A) cytosolic and mitochondrial cytochrome c detection, (B) cytosolic cytochrome c levels, (C) mitochondrial cytochrome c levels, and (D) caspase-3 activity. Cytochrome c levels were detected by Western blotting. Lanes 1–3 = control; Lanes 4–7 = ethanol; Lanes 8–10 = betaine; Lanes 11–13 = betaine and ethanol. Caspase-3 activity in 25 µg of cytosolic protein was determined based on the cleavage of Ac-DEVD-AFC (3 mg/mL) to AFC. Values are expressed as mean ± SEM (n = 3–4). *P < 0.05 compared to control.
Caspase-3 is one of the final effector caspases in the apoptosis pathway (along with caspase-6 and caspase-7). Therefore, caspase-3 levels were assayed to determine if the cytochrome c release into the cytosol resulted in apoptosis and if betaine could prevent this release. However, ethanol treatment alone led to a 19% decrease in caspase-3 activity. No changes were observed in either betaine alone or betaine followed by ethanol groups (Figure 1D).
Oxidative damage
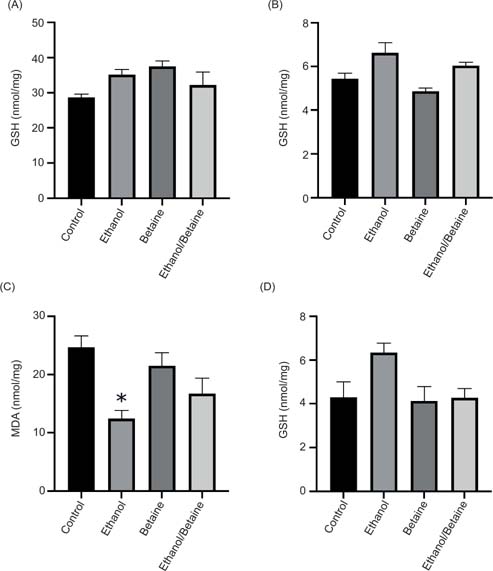
GSH levels were generally increased in ethanol and betaine supplementation. Ethanol treatment and betaine supplementation alone led to a 23 and 31% increase in cytosolic GSH levels, respectively (Figure 2A). A similar pattern occurred with mitochondrial GSH levels where ethanol caused an increase of 22% in mitochondrial GSH levels, and betaine followed by ethanol resulted in a 24% increase (Figure 2B). To assess whether betaine supplementation provided oxidative protective properties to the liver, MDA levels were assessed as a parameter for lipid peroxidation. Surprisingly, ethanol treatment led to a 50% reduction in cytosolic MDA levels (P < 0.05). Betaine alone had little effect (13% decrease), whereas betaine followed by ethanol caused a 22% reduction (Figure 2C). In the mitochondria, ethanol caused a 47% increase in MDA formation (Figure 2D). Betaine alone as well as the betaine and ethanol group also showed no changes.
Figure 2: The effect of betaine on ethanol-induced changes in (A) cytosolic GSH levels, (B) mitochondrial GSH levels, (C) cytosolic MDA formation, and (D) mitochondrial MDA formation. The “betaine” experimental animals were given drinking water containing 2% betaine (w/v) for 6 days. Saline (0.15 mol/L NaCl) or ethanol (75 mmol/kg bodyweight) was administered i.p. on day 7, and 24 h later the animals were sacrificed. Liver mitochondrial and cytosolic fractions were prepared as described in the methods. Values are expressed as mean ± SEM (n = 3–4). *P < 0.05 compared to control.
Antioxidant enzyme activity
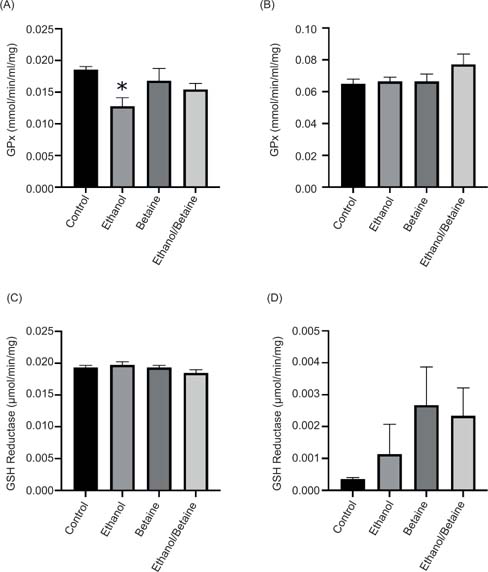
Ethanol exposure alone led to a 31% decrease (P < 0.05) in cytosolic GPx activity when compared to the control (Figure 3A). Both the betaine only and ethanol- and betaine-treated groups led to a 10% and an 8% decrease, respectively, when compared to the corresponding controls (Figure 3B). In the mitochondria, no changes were seen in the ethanol and betaine-only groups. However, in the ethanol- and betaine-treated group, a 16% increase was observed (Figure 3B). GR catalyzes the reduction of GSSG to reduced GSH. In the cytoplasm, no changes were observed in GR activity in any treatment group (Figure 3C). On the other hand, in the mitochondria, GR was increased by 175 and 575% following treatment with ethanol and betaine supplementation, respectively (Figure 3D). In the ethanol- and betaine-treated group, a 15% reduction was observed when compared to the betaine only group (Figure 3D).
Figure 3: The effect of betaine on ethanol-induced changes in (A) cytosolic GPx activity, (B) mitochondrial GPx activity, (C) cytosolic GSH reductase activity, and (D) mitochondrial GSH reductase activity. Following treatment, liver mitochondrial and cytosolic fractions were prepared as previously described, and the antioxidant enzyme activity was determined as described in the methods. Values are expressed as mean ± SEM (n = 3–4). *P < 0.05 compared to control.
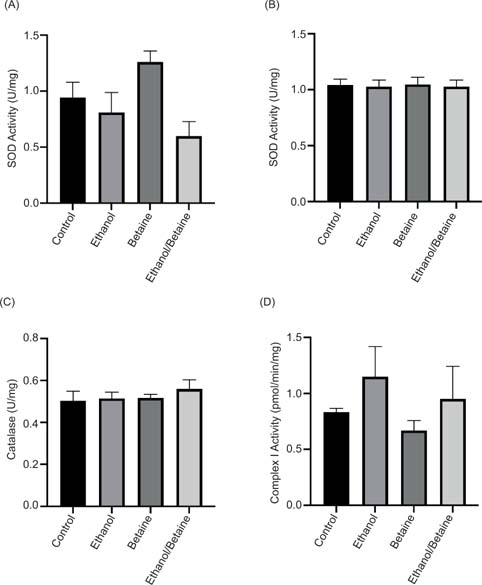
In the cytoplasm, ethanol treatment alone led to a 14% decrease in SOD activity, whereas in contrast, betaine treatment alone led to a 34% increase (Figure 4A). Ethanol and betaine treatment led to a 53% reduction in SOD activity in the cytoplasm when compared to the betaine control (Figure 4A). In the mitochondria, no changes to SOD activity were observed across all treatment groups (Figure 4B). The activity of catalase was also measured to assess its function. No changes in catalase activity were observed in either the ethanol alone, betaine alone, or a combination of ethanol and betaine treatments (Figure 4C).
Figure 4: The effect of betaine on ethanol-induced changes in (A) cytosolic SOD activity, (B) mitochondrial SOD, (C) catalase activity, and (D) complex I activity. Following treatment, liver mitochondrial and cytosolic fractions were prepared as previously described and the antioxidant enzyme activity was determined as described in the methods. Values are expressed as mean ± SEM (n = 3–4).
Ethanol treatment alone led to a 50% increase in complex I activity and betaine supplementation alone led to a 13% decrease in complex I activity, respectively (Figure 4D). Betaine treatment followed by ethanol led to a 43% increase in complex I activity when compared to the betaine only group (Figure 4D).
Discussion
To date, there have been few studies assessing the effects of both ethanol and betaine on oxidative damage and apoptosis in adolescents. Therefore, we investigated how ethanol affects juvenile hepatic cells through changes in oxidative damage, apoptosis, and liver function, and whether supplementation of betaine can provide protection or minimize the effects of oxidative liver injury.
The administration of ethanol caused a rise in the levels of hepatic enzymes, indicating structural membrane damage to the hepatocytes. Betaine was unable to prevent the rise in ALT levels following ethanol exposure (Table 2), although, it is perhaps the length of betaine treatment or concomitant betaine treatment that is essential for protection. In animal studies, prolonged betaine treatment has been shown to be efficacious in ameliorating the adverse effects of acute ethanol or lipopolysaccharide challenge (19, 20, 29, 30). Ethanol also led to a slight decrease in protein synthesis as measured by albumin, which may be due to perturbed elongation factors reducing protein translation (31). Pretreatment with betaine normalized protein and albumin levels (Table 2), which is consistent with previous findings (32).
Cytochrome c levels were significantly raised in the cytoplasm following ethanol treatment, and the levels were consequently decreased in the mitochondria. Destabilization of the cardiolipin anchor may have occurred due to oxidative stress releasing cytochrome c into the cytosol. Betaine pretreatment maintained the levels of mitochondrial cytochrome c and reduced the release of cytochrome c into the cytosol (Figure 1). Betaine prevents adenosine-induced apoptosis by preventing increases in intracellular S-Adenosylmethionine (SAM) levels in rat hepatocytes (21). SAM supplementation has also shown to protect against apoptosis in rat hepatocytes after 24-h incubation with ethanol (33). This suggests that induction of the methionine-homocysteine recycling mechanism reduces cytochrome c release, which may in part be mediated by oxidative stress. However, this did not translate into an increase in caspase-3 activity, which is an ATP-dependent activation step (34). Since ATP levels drop following ethanol metabolism (35–37) or can be depleted following 24-h starvation (38) in conjunction with oxidative damage (39, 40), this may explain the lack of caspase 3 activity.
Ethanol reduces the level of GSH (41–43); however, in our studies, cytosolic and mitochondrial GSH levels were generally maintained or slightly increased (Figure 2), and MDA levels were generally correspondingly decreased (Figure 2). These findings suggest that compensatory mechanisms are involved in preventing oxidative damage in response to acute ethanol administration (42, 44). This may be due to an increased synthesis of GSH in the cytoplasm along with its subsequent import to the mitochondria or increased synthesis of antioxidant enzymes. This shows that the liver can respond to an acute ethanol dose and that the increased GSH production offers protection against basal oxidative damage.
Cytosolic GSH levels were only slightly reduced with betaine and ethanol treatments (Figure 2), which may explain the corresponding lower cytosolic MDA levels, indicating that betaine is conferring some protection, perhaps via utilization of cytosolic GSH. A similar pattern was also observed in the mitochondria. The ameliorative effect of betaine on hepatic GSH and MDA levels has been shown in both acute and chronic models of ethanol intake (19, 20). Our data support the protective effects of betaine on GSH levels; however, the mechanism causing the increase remains to be elicited.
Ethanol consumption affects mitochondrial morphology, which alters the capacity of oxidative phosphorylation (18, 45). Chronic treatment of ethanol induces loss of oxidation phosphorylation proteins, which can be prevented by betaine supplementation (15). However, here we found complex I activity increased after both acute ethanol treatment and ethanol and betaine combined treatment. Further studies are required to explore this finding.
Chronic ethanol treatment leads to decreases in both cytosolic and mitochondrial GPx (46). Concordantly, we demonstrated acute ethanol exposure led to a significant decrease in GPx activity in the cytoplasm as well as in the betaine-treated groups, indicating decreased clearance of H2O2. GPx and catalase activity remained unchanged in the mitochondria across all treatment groups. Treatment with ethanol alone and treatment with both ethanol and betaine led to decreased SOD activity. The reduction in SOD activity may be due to ROS-induced enzyme degradation as well as a reduced synthesis of enzymes (47). However, as SOD activity increased in the cytoplasm with betaine treatment alone, betaine may provide some protection to oxidative damage via an increase in antioxidant enzymes. This demonstrates that the antioxidant enzymes are generally maintained after acute ethanol treatment in juvenile rats. Antioxidant enzymes are reduced with age and ethanol consumption (48–50), which may account for the higher antioxidant capacity in juvenile rats.
Conclusion
We have demonstrated that the livers from juvenile rats react differently to aged animals in response to acute ethanol by increasing GSH levels. Whilst the mechanism remains unclear, increasing GSH levels following acute ethanol could be due to higher basal levels and the capacity of antioxidant enzymes in adolescents. Betaine pretreatment was able to ameliorate or prevent some oxidative changes. Thus, betaine may serve as an effective therapeutic in the treatment of liver disease, as well as other diseases associated with oxidative damage.
Acknowledgments
Lucy Petagine and Hannah Everitt were supported by a scholarship from the University of Westminster.
REFERENCES
1. Mathurin P, Hadengue A, Bataller R, Addolorato G, Burra P, Burt A, et al. EASL clinical practical guidelines: Management of alcoholic liver disease. J Hepatol. 2012;57:399–420. 10.1016/j.jhep.2012.04.004
2. Chung T, Creswell KG, Bachrach R, Clark DB, Martin CS. Adolescent binge drinking. Alcohol Res Curr Rev. 2018;39:5–15.
3. Hibell B, Stergar E, Dernovšček Hafner N. The 2011 ESPAD report: Substance use among students in 36 European countries. The Swedish Council for Information on Alcohol and other Drugs. 2012. http://www.can.se/PageFiles/2619/The_2011_ESPAD_Report_FULL.pdf?epslanguage=sv
4. Hibell B, Andersson B, Bjarnason T, Ahlström S, Balakireva O, Kokkevi A, et al. The ESPAD report 2003: Alcohol and other drug use among students in 35 European countries. The Swedish Council for Information on Alcohol and other Drugs. 2004.
5. Clark Goings T, Salas-Wright CP, Belgrave FZ, Nelson EJ, Harezlak J, Vaughn MG. Trends in binge drinking and alcohol abstention among adolescents in the US, 2002–2016. Drug Alcohol Depend. 2019;200:115–123. 10.1016/j.drugalcdep.2019.02.034
6. Tapia-Rojas C, Torres AK, Quintanilla RA. Adolescence binge alcohol consumption induces hippocampal mitochondrial impairment that persists during the adulthood. Neuroscience. 2019;406:356–68. 10.1016/j.neuroscience.2019.03.018
7. Bailey SM, Cunningham CC. Acute and chronic ethanol increases reactive oxygen species generation and decreases viability in fresh, isolated rat hepatocytes. Hepatology. 1998;28:1318–26. 10.1002/hep.510280521
8. Bailey SM, Pietsch EC, Cunningham CC. Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III. Free Radic Biol Med. 1999;27:891–900. 10.1016/S0891-5849(99)00138-0
9. Ismail NA, Okasha SH, Dhawan A, Abdel-Rahman AO, Shaker OG, Sadik NA. Antioxidant enzyme activities in hepatic tissue from children with chronic cholestatic liver disease. Saudi J Gastroenterol. 2010;16:90–4. 10.4103/1319-3767.61234
10. Chrobot AM, Szaflarska-Szczepanik A, Drewa G. Antioxidant defense in children with chronic viral hepatitis B and C. Med Sci Monit Int Med J Exp Clin Res. 2000;6:713–18. PMid: 11208397
11. Moreno I, Pichardo S, Jos A, Gómez-Amores L, Mate A, Vazquez CM, et al. Antioxidant enzyme activity and lipid peroxidation in liver and kidney of rats exposed to microcystin-LR administered intraperitoneally. Toxicon. 2005;45:395–402. 10.1016/j.toxicon.2004.11.001
12. Kurutas EB. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr J. 2016;15:71. 10.1186/s12937-016-0186-5
13. Purohit V, Abdelmalek MF, Barve S, Benevenga NJ, Halsted CH, Kaplowitz N, et al. Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: Summary of a symposium. Am J Clin Nutr. 2007;86:14–24. 10.1093/ajcn/86.1.14
14. Barak AJ, Beckenhauer HC, Tuma DJ. Betaine effects on hepatic methionine metabolism elicited by short-term ethanol feeding. Alcohol. 1996;13:483–486. 10.1016/0741-8329(96)00040-7
15. Kharbanda KK, Todero SL, King AL, Osna NA, McVicker BL, Tuma DJ, et al. Betaine treatment attenuates chronic ethanol-induced hepatic steatosis and alterations to the mitochondrial respiratory chain proteome. Int J Hepatol. 2012;2012:962183. 10.1155/2012/962183
16. Barak AJ, Beckenhauer HC, Mailliard ME, Kharbanda KK, Tuma DJ. Betaine lowers elevated S-adenosylhomocysteine levels in hepatocytes from ethanol-fed rats. J Nutr. 2003;133:2845–8. 10.1093/jn/133.9.2845
17. Kanbak G, İnal M, Bayçu C. Ethanol-induced hepatotoxicity and protective effect of betaine. Cell Biochem Funct. 2001;19:281–5. 10.1002/cbf.926
18. Kharbanda KK, Mailliard ME, Baldwin CR, Beckenhauer HC, Sorrell MF, Tuma DJ. Betaine attenuates alcoholic steatosis by restoring phosphatidylcholine generation via the phosphatidylethanolamine methyltransferase pathway. J Hepatol. 2007;46:314–21. 10.1016/j.jhep.2006.08.024
19. Kim SJ, Jung YS, Kwon DY, Kim YC. Alleviation of acute ethanol-induced liver injury and impaired metabolomics of S-containing substances by betaine supplementation. Biochem Biophys Res Commun. 2008;368:893–8. 10.1016/j.bbrc.2008.02.003
20. Balkan J, Öztezcan S, Küçük M, Çevikbas¸ U, Koçak-Toker N, Uysal M. The effect of betaine treatment on triglyceride levels and oxidative stress in the liver of ethanol-treated guinea pigs. Exp Toxicol Pathol. 2004;55:505–9. 10.1078/0940-2993-00347
21. Kharbanda KK, Rogers II DD, Mailliard ME, Siford GL, Barak AJ, Beckenhauer HC, et al. A comparison of the effects of betaine and S-adenosylmethionine on ethanol-induced changes in methionine metabolism and steatosis in rat hepatocytes. J Nutr. 2005;135:519–24. 10.1093/jn/135.3.519
22. Kharbanda KK, Todero SL, Ward BW, Cannella JJ, Tuma DJ. Betaine administration corrects ethanol-induced defective VLDL secretion. Mol Cell Biochem. 2009;327:75–8. 10.1007/s11010-009-0044-2
23. Alirezaei M, Jelodar G, Niknam P, Ghayemi Z, Nazifi S. Betaine prevents ethanol-induced oxidative stress and reduces total homocysteine in the rat cerebellum. J Physiol Biochem. 2011;67:605–12. 10.1007/s13105-011-0107-1
24. Patel VB, Spencer CH, Young TA, Lively MO, Cunningham CC. Effects of 4-hydroxynonenal on mitochondrial 3-hydroxy-3-methylglutaryl (HMG-CoA) synthase. Free Radic Biol Med. 2007;43:1499–507. 10.1016/j.freeradbiomed.2007.08.004
25. Preedy VR, Gove CD, Panos MZ, Sherwood R, Portmann B, Williams R, et al. Liver histology, blood biochemistry and RNA, DNA and subcellular protein composition of various skeletal muscles of rats with experimental cirrhosis: Implications for alcoholic muscle disease. Alcohol Alcohol. 1990;25:641–9. 10.1093/oxfordjournals.alcalc.a045061
26. Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: Applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–22. 10.1016/0003-2697(69)90064-5
27. Bar-Or D, Rael LT, Lau EP, Rao NKR, Thomas GW, Winkler JV, et al. An analog of the human albumin N-terminus (Asp-Ala-His-Lys) prevents formation of copper-induced reactive oxygen species. Biochem Biophys Res Commun. 2001;284:856–62. 10.1006/bbrc.2001.5042
28. Yemis¸ci M, Gürsoy-Özdemir Y, Caban S, Bodur E, Çapan Y, Dalkara T. Transport of a caspase inhibitor across the blood-brain barrier by chitosan nanoparticles. In: Methods in enzymology. Editor, Nejat Düzgünes¸ Academic Press; Netherlands:Elsevier. 2012. p. 253–69.
29. Kim SK, Kim YC. Attenuation of bacterial lipopolysaccharide-induced hepatotoxicity by betaine or taurine in rats. Food Chem Toxicol. 2002;40:545–9. 10.1016/S0278-6915(01)00102-8
30. Erman F, Balkan J, Çevikbas¸ U, Koçak-Toker N, Uysal M. Betaine or taurine administration prevents fibrosis and lipid peroxidation induced by rat liver by ethanol plus carbon tetrachloride intoxication. Amino Acids. 2004;27:199–205. 10.1007/s00726-004-0105-5
31. Lang CH, Liu X, Nystrom G, Wu D, Cooney RN, Frost RA. Acute effects of growth hormone in alcohol-fed rats. Alcohol Alcohol. 2000;35:148–58. 10.1093/alcalc/35.2.148
32. Hayes KC, Pronczuk A, Cook MW, Robbins MC. Betaine in sub-acute and sub-chronic rat studies. Food Chem Toxicol. 2003;41:1685–700. 10.1016/S0278-6915(03)00196-0
33. del Pilar Cabrales-Romero M, Márquez-Rosado L, Fattel-Fazenda S, Trejo-Solís C, Arce-Popoca E, Alemán-Lazarini L, et al. S-adenosyl-methionine decreases ethanol-induced apoptosis in primary hepatocyte cultures by a c-Jun N-terminal kinase activity-independent mechanism. World J Gastroenterol. 2006;12:1895–904. 10.3748/wjg.v12.i12.1895
34. Ferrari D, Stepczynska A, Los M, Wesselborg S, Schulze-Osthoff K. Differential regulation and ATP requirement for caspase-8 and caspase-3 activation during CD95-and anticancer drug-induced apoptosis. J Exp Med. 1998;188:979–84. 10.1084/jem.188.5.979
35. Young TA, Bailey SM, Van Horn C, Cunningham CC. Chronic ethanol consumption decreases mitochondrial and glycolytic production of ATP in liver. Alcohol Alcohol. 2006;41:254–60. 10.1093/alcalc/agl017
36. Nassir F, Ibdah JA. Role of mitochondria in alcoholic liver disease. World J Gastroenterol. 2014;20:2136–42. 10.3748/wjg.v20.i9.2136
37. Andreoli SP. Mechanisms of endothelial cell ATP depletion after oxidant injury. Pediatr Res. 1989;25:97–101. 10.1203/00006450-198901000-00021
38. Ghanbari-Niaki A, Désy F, Lavoie JM. Effects of phosphate injection on metabolic and hormonal responses to exercise in fructose-injected rats. Physiol Behav. 1999;67:747–52. 10.1016/S0031-9384(99)00130-4
39. Crompton M. The mitochondrial permeabitity transition pore and its role in cell death. Biochem J. 1999;341:233–49. 10.1042/bj3410233
40. Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis. 2007;12:815–33. 10.1007/s10495-007-0723-y
41. Cederbaum AI. Microsomal generation of reactive oxygen species and their possible role in alcohol hepatotoxicity. Alcohol Alcohol Suppl. 1991;1:291–6.
42. Vogt BL, Richie JP. Glutathione depletion and recovery after acute ethanol administration in the aging mouse. Biochem Pharmacol. 2007;73:1613–21. 10.1016/j.bcp.2007.01.033
43. Kade S, Herzog N, Schmidtke K-U, Küpper J-H. Chronic ethanol treatment depletes glutathione regeneration capacity in hepatoma cell line HepG2. J Cell Biotechnol. 2016;1:183–90. 10.3233/JCB-15019
44. Guerri C, Grisolía S. Changes in glutathione in acute and chronic alcohol intoxication. Pharmacol Biochem Behav. 1980;13:53–61. 10.1016/S0091-3057(80)80009-8
45. Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology. 2002;122:2049–63. 10.1053/gast.2002.33613
46. Bailey SM, Patel VB, Young TA, Asayama K, Cunningham CC. Chronic ethanol consumption alters the glutathione/glutathione peroxidase-1 system and protein oxidation status in rat liver. Alcohol Clin Exp Res. 2001;25:726–33. 10.1111/j.1530-0277.2001.tb02273.x
47. Sriram KI, Lakshmi CJ. Endurance exercise-induced alterations in antioxidant enzymes of old albino male rats. Curr Sci. 2001;80:921–3.
48. Mallikarjuna K, Shanmugam KR, Nishanth K, Wu MC, Hou CW, Kuo CH, et al. Alcohol-induced deterioration in primary antioxidant and glutathione family enzymes reversed by exercise training in the liver of old rats. Alcohol. 2010;44:523–9. 10.1016/j.alcohol.2010.07.004
49. Cahill A, Hershman S, Davies A, Sykora P. Ethanol feeding enhances age-related deterioration of the rat hepatic mitochondrion. Am J Physiol Liver Physiol. 2005;289:G1115–23. 10.1152/ajpgi.00193.2005
50. Meier P, Seitz HK. Age, alcohol metabolism and liver disease. Curr Opin Clin Nutr Metab Care. 2008;11:21–6. 10.1097/MCO.0b013e3282f30564